

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cardiovascular diseases are the leading cause of death worldwide, and an estimated 17.9 million people died from cardiovascular diseases in 2019 alone, responsible for 32% of all deaths.1) While the prevalence of coronary artery disease has shown a downward trend,2) heart failure (HF) has continued to increase in prevalence.3) In contrast to the dramatic improvement of the mortality rates in patients with coronary artery disease and valvular heart disease, HF has displayed the opposite trend.3) Traditionally, HF was thought to originate from reduced ejection fraction (HFrEF) caused by myocardial infarction, myocarditis, or cardiomyopathy, etc. The counterpart, HF with preserved EF (HFpEF) was first described in 1982 as HF in older patients (>75 years old), with a somewhat better prognosis than HFrEF.4) Once much less common than HFrEF, HFpEF now accounts for approximately half of all HF patients due to an aging population with hypertension, diabetes mellitus, and obesity.3) The prognosis of HFpEF is currently known to be as dismal as HFrEF, with a 75% mortality within 5 years regardless of EF.5) HFpEF remains a challenging clinical entity which is resistant to routine medical management. Tens of clinical trials have attempted to apply medications adapted from HFrEF (beta-blockers, angiotensin converting enzyme inhibitors, angiotensin receptor blockers, mineralocorticoid antagonists, angiotensin receptor-neprilysin inhibitors, and sodium-glucose cotransporter-2 inhibitors), but none of them have shown a strong survival benefit among patients with EF ≥50%.6)7)8)9)10)11) Sudden death is the most common mode of death in patients with HFpEF,12) but the underlying mechanisms remain unknown. Although ventricular arrhythmias are responsible for the majority of sudden deaths,13) this has not been unquestionably proven in patients with HFpEF. Modern devices such as an implantable cardioverter-defibrillator are not routinely indicated for the primary prevention of sudden death, due to the normal EF in patients with HFpEF.14) Ventricular arrhythmias are commonly reported in patients with HFpEF, but the burden and mechanisms have not been well investigated. This review will focus on sudden death and mechanisms of ventricular arrhythmias in HFpEF.
Go to : 
SUDDEN DEATH IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
Sudden death
Sudden death is defined as “death occurring less than 24 hours from onset of symptoms” according to the 10th revision of International Classification of Disease. However, in clinical studies, the definition has been used more strictly as death occurring less than 1 hour from onset of symptoms.14) World Health Organization defines sudden death as death occurring less than 1 hour from acute changes in witnessed cases, or if a patient is found dead within 24 hours in unwitnessed cases.15) Based on the etiology, sudden death can be divided into sudden cardiac death (SCD) and sudden non-cardiac death (SNCD). SCD is described as the unexpected death from a cardiac cause within 1 hour from the onset of symptoms.14) Sudden cardiac arrest (SCA) is interchangeably used with SCD, but SCA does not necessarily result in SCD. SNCD encompasses non-cardiac causes of death such as pulmonary disease (40%), infectious disease (20%), cerebrovascular disease (18%), and neurologic diseases (8%) etc.15) The investigation of sudden death is challenging due to the unexpected nature of the event. Retrospective death certificate-based investigation has the propensity to overestimate incidence, almost 3-fold compared to a prospective approach.16) The prospective, multiple source surveillance using emergency medical services, medical examiners, and hospitals or providers, is feasible and more accurate than retrospective investigations.16) Furthermore, prospective autopsy investigation can accurately estimate the burden of sudden death.17)
Sudden cardiac death
Although non-cardiac deaths can be sudden, most sudden deaths (up to 90%) are cardiac18); hence sudden death and SCD are used interchangeably. Anatomical and/or functional substrates interplay with initiating the event, promoting the maintenance of electrical instability. In patients with advanced HFrEF, ventricular arrhythmias were the most common cause of cardiac arrest, comprising 52% of cases.19) SCD is often challenging to investigate since approximately 40% of cases are unwitnessed.20) The most valuable clue (not the evident cause) can be obtained from autopsy.17) The standard procedure of autopsy in cases of sudden death is: exclusion of non-cardiovascular causes (cerebral hemorrhage, asthma or pneumonia, ruptured aortic aneurysm etc.), histology and imaging of hearts, and toxicology and genetic testing of hearts.21)
Cause of death vs. Mode of death
Although interchangeably used in most literature, cause of death and mode of death are somewhat different and should be used separately. Cause of death is defined as the disease or injury that initiated the train of morbid events leading directly to death.22) The common cause of deaths are heart disease, malignant neoplasms, chronic lower respiratory diseases, accidents, cerebrovascular diseases, etc.23) The mode of death is usually used in the mortality analysis of HF, as sudden death is the most common entity in which the cause is largely unknown. Mode of death can be divided into cardiovascular and non-cardiovascular categories. Common modes of cardiovascular deaths are sudden death, heart failure, myocardial infarction and cerebrovascular accident, etc.24) Non-cardiovascular death can be sudden as well, but is more commonly non-sudden due to pulmonary disease, neoplasm, infectious disease, renal disease and neurologic disease.
Risk factors of sudden death
Coronary artery disease is the most common risk factor for sudden death, and approximately 80% of sudden death cases have coronary artery disease as the underlying substrate.18) Autopsy studies show that occlusive coronary artery thrombus was found in 15–64% of sudden death patients.25) Cardiomyopathy represents the second largest risk factor for sudden death. Hypertrophic cardiomyopathy, dilated cardiomyopathy, and arrhythmogenic right ventricular dysplasia are common presentations. Reduced EF is a major independent predictor of sudden death in patients with ischemic and nonischemic cardiomyopathy.19) In patients with severe HFrEF, non-sustained ventricular tachycardia (NSVT) was shown to be an independent risk factor of sudden death (2.7-fold).26) Left ventricular hypertrophy resulting from hypertension, valvular heart disease, or congenital heart disease, represents a significant risk factor for sudden death as well.27) Primary electrophysiological abnormalities, including channelopathy, are also important risk factors of sudden death.
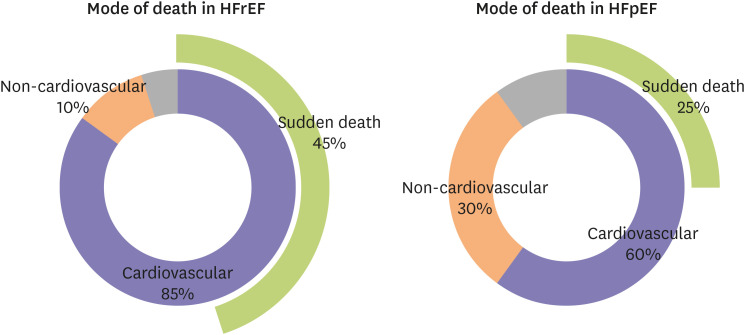
Mode of death in heart failure with preserved ejection fraction
The mode of death in patients with HFpEF was investigated in various randomized controlled trials and prospective registries. Cardiovascular deaths were responsible for 60% of all deaths in patients with HFpEF, while non-cardiovascular deaths comprised 30% of all deaths (Figure 1). Sudden death was the most common mode of death, consisting of roughly 25% of all death in patients with HFpEF (Figure 1). This mode of death in patients with HFpEF was somewhat different from that of HFrEF patients: there was less burden of cardiovascular mode of death, and reduced sudden deaths in patients with HFpEF. According to the Irbesartan in Heart Failure With Preserved Ejection Fraction (I-PRESERVE, 4,128 HFpEF patients) trial, 60% of the deaths of HFpEF patients were cardiovascular, and 30% were non-cardiovascular (10% unknown).24) Among the cardiovascular deaths, sudden death was the most common mode of death in HFpEF; it accounted for 26% of all deaths (231/881 deaths), followed by heart failure (14%), cerebrovascular accident (9%), and myocardial infarction (5%).24) Non-cardiovascular modes of death comprise 30% of all death in HFpEF.24) In the Aldosterone Antagonist Therapy for Adults With Heart Failure and Preserved Systolic Function (TOPCAT, 3,445 HFpEF patients) trial, 63.9% of all deaths in HFpEF patients were cardiovascular and 27.6% were non-cardiovascular.9) Sudden death was the most common mode of death, accounting for 24.3% of all deaths (128/526 deaths), followed by heart failure (12.7%), myocardial infarction (6.3%), and stroke (4.8%).9) Non-cardiovascular causes of death included malignancy (9.9%), infection (7.0%), gastrointestinal (3.2%) and pulmonary (2.7%).9) The Japanese Cardiac Registry of Heart Failure in Cardiology (JCARE-CARD) was a prospective registry which looked at the mode of death in 323 patients hospitalized with worsening HF (including 169 patients with HFpEF).28) Similar to the trend displayed in randomized controlled trials, cardiovascular death was responsible for 58% of deaths, while 28% of deaths were due to a non-cardiovascular mode.28) Contrary to the randomized controlled trials, the most common mode of death was worsening HF (59/169 deaths, 35%) and sudden death only accounted for 11% of deaths (18/169 deaths).28) There have been a few more registries and epidemiological studies,12) but the number of patients who participated was low, hence a meaningful conclusion could not be drawn.
Predictors of sudden death in heart failure with preserved ejection fraction
For patients with HFrEF, reduced EF (less than 30–35%) implies an increased risk of arrhythmia-related sudden death.29)30)31) NSVT is also an independent marker for increased sudden death (2.7-fold) in patients with HFrEF as compared to HFrEF patients without NSVT.26) Whereas the predictors of sudden death are well studied in patients with HFrEF, little is known about the risk factors that can predict sudden death in HFpEF. Adabag et al.32) performed a post-hoc analysis of the I-PRESERVE trial in order to develop a prediction model of sudden death. A multivariable Cox regression model was developed based on age, male gender, history of diabetes mellitus and myocardial infarction, left bundle branch block on ECG, and the natural logarithm of NT-proBNP, which identified patients with HFpEF who had a risk of sudden death over 5 years.32) Vaduganathan et al.33) investigated predictors of sudden death in patients with HFpEF using TOPCAT trial data, and it was found that male sex and insulin treated diabetes mellitus both independently predicted an increased risk of sudden death in HFpEF patients. Although echocardiogram and cardiac magnetic resonance imaging have been used to predict the risk of sudden death in patients with HFrEF, there is no study which used imaging studies to predict sudden death risk in patients with HFpEF.
Causes of sudden death in heart failure with preserved ejection fraction
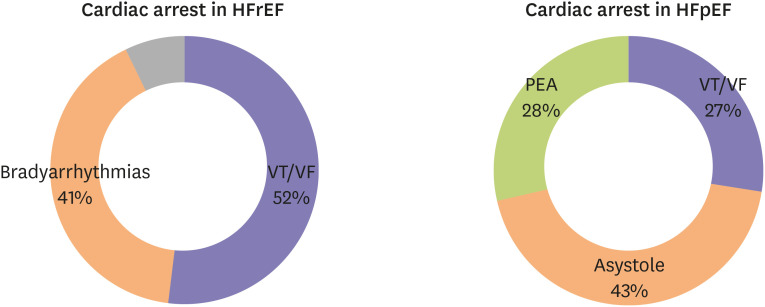
The role of ventricular arrhythmias in sudden deaths of HFpEF was investigated in a pre-clinical model of HFpEF. In a Dahl salt-sensitive rat model of HEpEF, sudden deaths were probed with ambulatory ECG monitoring.34) Cho et al.34) discovered that 75% of sudden deaths in HFpEF rats are due to ventricular arrhythmias. As pointed out previously, the cause of sudden death is largely unknown in HFpEF patients, even with extensive investigations and autopsies. In hospitalized patients with HFrEF, ventricular arrhythmias (ventricular tachycardia [VT] or fibrillation [VF]) were responsible for 52% of cardiac arrest cases, while brady-arrhythmias were present in 41% of cases (Figure 2).19) Although sudden death can be largely attributed to ventricular arrhythmias, there is no evidence to support this assumption in patients with HFpEF. Woolcott et al. investigated the prevalence of shockable rhythms (VT or VF) at the time of cardiac arrest in HFpEF populations.35) Among 282 HFpEF patients with sudden cardiac arrest, only 27.0% of cases were shockable rhythms; asystole was present in 43.3% of cases, and pulseless electrical activity (PEA) in 28.7% (Figure 2).35) The findings here suggest that in contrast to sudden death in HFrEF, in which ventricular arrhythmias are the major cause of death, ventricular arrhythmias might play a less significant role in sudden death among HFpEF patients.
Go to :
VENTRICULAR ARRHYTHMIAS IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
There have been several recent studies which investigated the burden of ventricular arrhythmias in patients with HFpEF. Gutierrez et al.36) performed a retrospective review of HFpEF patients who had ambulatory ECG monitoring or pacemaker interrogations. NSVT was found in 32.5% of HFpEF patients who underwent ambulatory ECG monitoring, and 44.7% of HFpEF patients who had pacemaker interrogations.36) Most importantly, NSVT was associated with a 3.4-fold increased risk of death in HFpEF patients.36) A similar retrospective medical record review was done by Cho et al.37) for HFpEF patients who underwent ambulatory Zio patch monitoring. Similarly, NSVT was found in 37% of HFpEF patients, as compared to 16% occurrence in patients without HFpEF.37) Additionally, sustained VT was identified in 1/110 HFpEF patients.37) Covariate-adjusted logistic regression showed that HFpEF was associated with an increased risk of NSVT.37) Another important prospective observational study was done (VIP-HF study) in the Netherlands for patients with HFpEF using an implantable loop recorder.38) Eighteen percent of HFpEF patients were shown to have NSVT during the median follow-up of 657 days.38) Only 1 out of 113 patients developed sustained VT.38) In summary, NSVT was very frequent in patients with HFpEF (up to 35% of patients), but sustained VT was infrequent (up to 1%).
Go to :
MECHANISMS OF VENTRICULAR ARRHYTHMIAS IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
Hypertension and ventricular hypertrophy
Approximately 90% of patients with HFpEF display hypertension as an underlying risk factor,10)11) and furthermore, hypertension has been regarded as the key pathologic risk factor in the traditional model of HFpEF.39) Systemic hypertension causes a pressure overload of the ventricle and leads to concentric hypertrophy. Approximately 30–40% of patients with HFpEF show evidence of left ventricular hypertrophy.8)9) Patients with hypertension and left ventricular hypertrophy are more prone to develop ventricular ectopy and ventricular arrhythmias when compared to hypertensive patients without hypertrophy.40)41)42) In patients with decompensated hypertrophy, connexin 43 expression is diminished and its distribution becomes heterogeneous.43) Decreased expression and heterogeneous distribution of connexin 43 can lead to a slow conduction velocity,44) which can act as a substrate of re-entry (Figure 3). Reduced conduction velocity due to phosphorylation and lateralization of connexin 43 has also been implicated in the arrhythmogenesis in HF.44) Since HFpEF is partly caused by hypertension-induced ventricular hypertrophy, connexin 43 reduction and redistribution are important substrates of ventricular arrhythmias in HFpEF.

Delayed repolarization
Delayed repolarization has been a recurrent theme in the pathogenesis of cardiac arrhythmias.45)46) Cho et al.37) retrospectively studied 110 HFpEF patients and compared them to 97 patients without HFpEF, and discovered that the QTc interval was prolonged in patients with HFpEF (451±33 ms) as compared to patients without HFpEF (432±29 ms). Wilcox et al.47) studied QTc prolongation in 75 patients with HF and discovered that the QTc interval was associated with diastolic dysfunction. The mechanisms of delayed repolarization were investigated in a Dahl salt-sensitive rat model of HFpEF by Cho et al.48) and it was revealed that the underlying mechanisms were the down-regulations of several potassium currents. The prolonged phase of action potential is vulnerable to the triggered activity such as early afterdepolarization (EAD),49) and EAD has been regarded as a trigger of ventricular arrhythmias (Figure 4).50)51) Among the various potassium currents, the transient outward potassium current (Ito) is arguably the major repolarizing current in animal and human cardiomyocytes.52)53)54)55) Cho et al.56) recently studied cardiac progenitor cells in a Dahl salt-sensitive rat model of HFpEF, and showed that the up-regulation of the Ito potassium current can mitigate the propensity for ventricular arrhythmias and prolong the survival of HFpEF rats.
 | Figure 4Delayed repolarization of HFpEF is caused by down-regulations of various potassium channels (transient outward and rapid component delayed rectifier potassium channels in this figure) and provides a window for EAD to initiate ventricular arrhythmias. Calcium leakeage caused by altered EC copling in HFpEF can trigger DAD and initiate ventricular arrhythmias.AP = action potential; DAD = delayed afterdepolarization; EAD = early afterdepolarization; HFpEF = heart failure with preserved ejection fraction.
|
Calcium homeostasis and excitation-contraction coupling
Alterations in intracellular calcium homeostasis which lead to abnormal excitation-contraction coupling (EC coupling) play an important role in ventricular arrhythmogenesis. Phosphorylation of the ryanodine receptor 2 (RyR2) by protein kinase A during increased adrenergic tone in HFrEF is known to dissociate FKBP12.6 from RyR2, hence increasing the open probability of the channel leading to defective calcium release during diastole.57) This calcium leakage causes sparks which can propagate throughout the cardiomyocyte and generate calcium waves, which can trigger delayed afterdepolarization (DAD).58) Kilfoil et al.59) studied EC coupling in Dahl salt-sensitive HFpEF rats and discovered that the L-type calcium current (ICaL) and calcium transient both increased in HFpEF cardiomyocytes as compared to normal cardiomyocytes. Systolic as well as diastolic intracellular calcium concentrations were also increased in HFpEF rat cardiomyocytes in comparison to normal cardiomyocytes.59) Furthermore, HFpEF cardiomyocytes exhibited an impaired beta-adrenergic response, i.e. a reduced increase in ICaL and calcium transient as compared to HFrEF cardiomyocytes.59) A significant increase in RyR2 phosphorylation at Ser2808 might play a role in calcium leakage from the sarcoplasmic reticulum.59) Frisk et al.60) studied different HFpEF rat models and revealed impaired diastolic calcium handling (reduced sarcoplasmic calcium uptake) was apparent in diabetic HFpEF rats, but not in hypertensive HFpEF rats. Runte et al.61) studied relaxation characteristics and calcium homeostasis in isolated myocardium in patients with HFpEF. In patients with HFpEF, isolated cardiomyocyte strips showed delayed relaxation and increased resting calcium levels, which is consistent with pre-clinical findings. Tachycardia-induced diastolic calcium levels were also increased in these cardiomyocyte strips, which potentially suggest increased calcium leaks.61) These intracellular diastolic calcium leaks from RyR2 can lead to ventricular arrhythmias by triggering DADs (Figure 4).62)
Inflammation and fibrosis
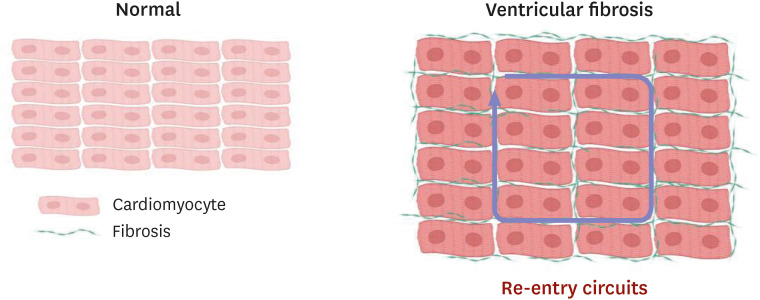
Traditionally, HFpEF has been attributed to underlying hypertension and ventricular hypertrophy, which can lead to myocardial stiffening and abnormal relaxation.63) The new paradigm shifts the focus from hypertension and ventricular hypertrophy to systemic inflammation and subsequent fibrosis.64)65) HFpEF is well known for its multiple co-morbidities (hypertension, diabetes mellitus, obesity, chronic kidney disease, pulmonary hypertension, atrial fibrillation, etc.), which contribute to increased systemic inflammation.66) Multiple inflammatory markers (interleukin [IL]-1, IL-6, tumor necrosis factor alpha, transforming growth factor beta [TGF-β], etc.) are proposed to mediate the inflammatory changes seen in HFpEF.65)66)67) Increased systemic inflammation and subsequent ventricular fibrosis are well described in rodent models of HFpEF.37)68)69) One of the important mediators of ventricular fibrosis is TGF-β, which activates fibroblasts to differentiate into myofibroblasts.70) These activated myofibroblasts produce collagen and deposit it in the extracellular space, contributing to cardiac fibrosis.66) Gallet et al.68) investigated cardiac progenitor cell therapy in a Dahl-salt sensitive rat model of HFpEF, and revealed that the TGF-β signaling pathway was involved in reduced cardiac fibrosis by cell therapy. Patients with HFpEF are shown to have increased myocardial fibrosis as well,71) and diffuse fibrosis in the ventricle is associated with slowing of wave propagation.72) Cardiac fibrosis provides an important substrate for cardiac arrhythmogenesis by yielding re-entry (Figure 5) and triggered activity such as EAD or DAD.73)
Go to :
CONCLUSION
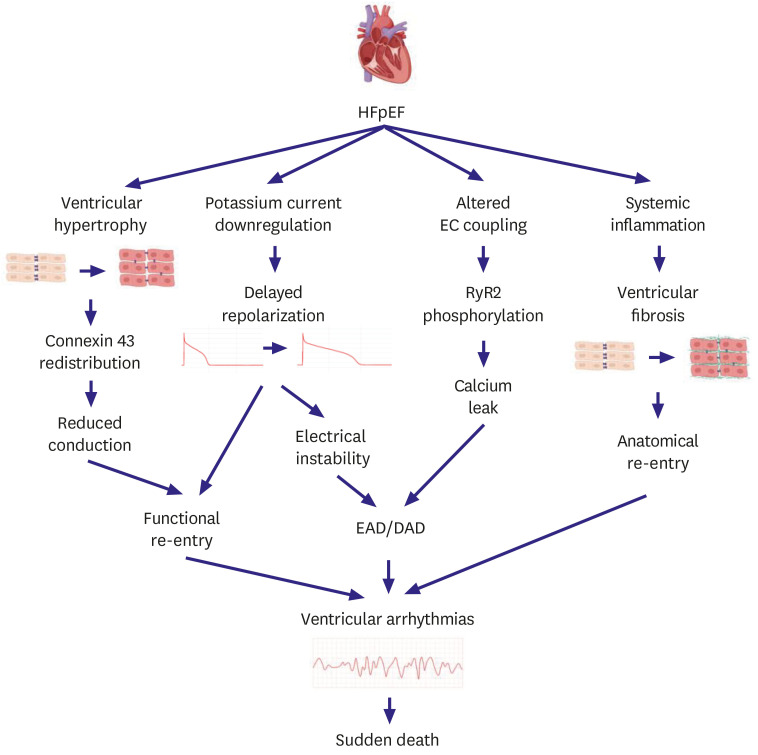
HFpEF accounts for approximately half of all heart failure cases. Sudden death is the most common mode of death in patients with HFpEF but its underlying mechanisms are not fully elucidated. Although ventricular arrhythmias are generally responsible for the majority of sudden deaths, their contribution to sudden deaths in HFpEF patients is likely less significant. The mechanisms of ventricular arrhythmias in HFpEF (Figure 6) are 1) reduced conduction velocity due to ventricular hypertrophy, 2) delayed repolarization due to potassium current down-regulation, 3) calcium leakage due to altered excitation-contraction coupling, and 4) increased ventricular fibrosis caused by systemic inflammation. Hypertension and subsequent ventricular hypertrophy reduce the conduction velocity in HFpEF hearts via heterogeneous distribution of connexin 43. Delayed repolarization caused by potassium current down-regulation in HFpEF hearts provides a window for early afterdepolarization to trigger ventricular arrhythmias. Altered excitation-contraction coupling in HFpEF can cause calcium to leak and trigger delayed afterdepolarization. Increased systemic inflammation and subsequent ventricular fibrosis provides functional re-entry substrates. Further research is warranted to investigate the detailed mechanisms of ventricular arrhythmias in HFpEF.
Go to :
 XML Download
XML Download