

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ependymomas are glial tumors that are thought to arise from primitive ependymal or subependymal cells in the vicinity of the ventricles and remnants of the central spinal canal38). Microscopically, ependymomas are moderately cellular gliomas corresponding to WHO grade 2 neoplasms and are well demarcated with a sharp tumor-parenchyma interface41). In aspect of intracranial ependymomas, so far the cure for this disease has eluded us and despite its histological benignancy, the majority of patients will die from their disease41). Although intracranial ependymoma is associated with an aggressive clinical course and poor prognosis, spinal ependymoma is linked with an indolent clinical course and good prognosis384246). The more difficult thing to estimate clinical course of ependymal tumors is that they are heterogeneous with regard to morphology, localization, and age at first clinical manifestation7). In children, 90% of ependymomas develop in the intracranial region and are associated with frequent recurrences18384650). In adults, 60% of ependymomas develop in the spinal cord, and recurrence is rare3850). The differences between intracranial and spinal ependymomas make the extrapolation of data from one to the other difficult50). Moreover, recent findings suggest that the histologic diagnosis of ependymomas may be insufficient for assigning an appropriate risk stratification strategy49).
Although intracranial and spinal ependymomas are histopathologically similar, their molecular biology is very heterogeneous, and they possess different DNA copy number alterations, messenger-RNA expression profiles, and genetic and epigenetic alterations as well as diverse transcriptional programs192427294046). The genetic landscape of ependymoma is also heterogeneous; these tumors show mostly complex aberration patterns with frequent deletions or gains of chromosomes, and the main losses occur on chromosomes 1p, 4q, 6q, 9, 10, 13q, 16, 17, 19q, 20q, and 22q161735474851). Some genetic researches revealed substantially different frequencies of genetic aberrations for different tumor locations7152144). Therefore, these results support the hypothesis that the histological entity "ependymoma" in fact comprises a group of related diseases that likely require different approaches and treatments172840). Furthermore, conventional therapies may fail to control tumor growth and progression due to the inherent heterogeneity of ependymoma, as demonstrated by analyses of their genetic and molecular anomalies1949). In this review, we describe the genetic differences between spinal ependymomas and their intracranial counterparts to better understand their prognosis. Research to improve our knowledge of the genetic differences between spinal and intracranial ependymomas will be essential to guide therapeutic strategies and estimate their prognoses.
Statistical analysis
For the statistical analyses, we used the R : A language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria) and the R-package "exact2×2" which is a Fishers exact test tool. If the statistical significance of a difference in genes, proteins, and chromosomal mutations was not described in each paper, we calculated the p-value using Fisher's exact 2 by 2 frequency probability to compare spinal ependymoma with its intracranial counterpart. For all tests, a level of p<0.05 was considered statistically significant.
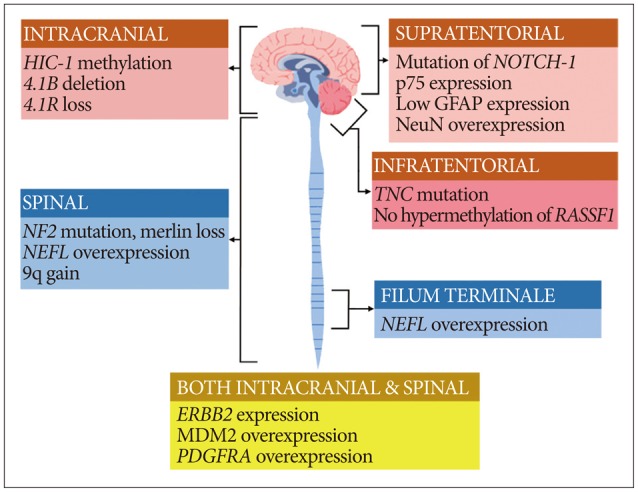
MOLECULAR GENETIC ABERRATIONS
Spinal cord ependymomas frequently exhibit mutations in NF2, NEFL overexpression, Merlin loss, and 9q gain, while intracranial ependymomas do not457123849). In addition, spinal cord ependymomas involve HES1, MYC, and GFAP overexpression1213). In myxopapillary ependymomas (MPEs), NEFL overexpression is frequently observed4). Intracranial ependymomas commonly display HIC-1 methylation, 4.1B deletion, and 4.1R loss3044. Supratentorial ependymoma display mutations in NOTCH-1, NeuN and p75 overexpression, and low expression of GFAP protein1213). Mutation in TNC, lack of hypermethylation in RASSF1A, and GFAP/NeuN expression may provide clues for the diagnosis of posterior fossa ependymoma121314). Although MEN1 and TP53 mutations have been rarely reported in ependymoma, they might be related to its recurrence or metastasis 5721). In turn, NEFL overexpression indicates a good prognosis and longer progression-free survival2). A graphical illustration of the key genes, proteins, and chromosomal aberrations related to the ependymoma subgroups according to tumor location is given in Fig. 1.
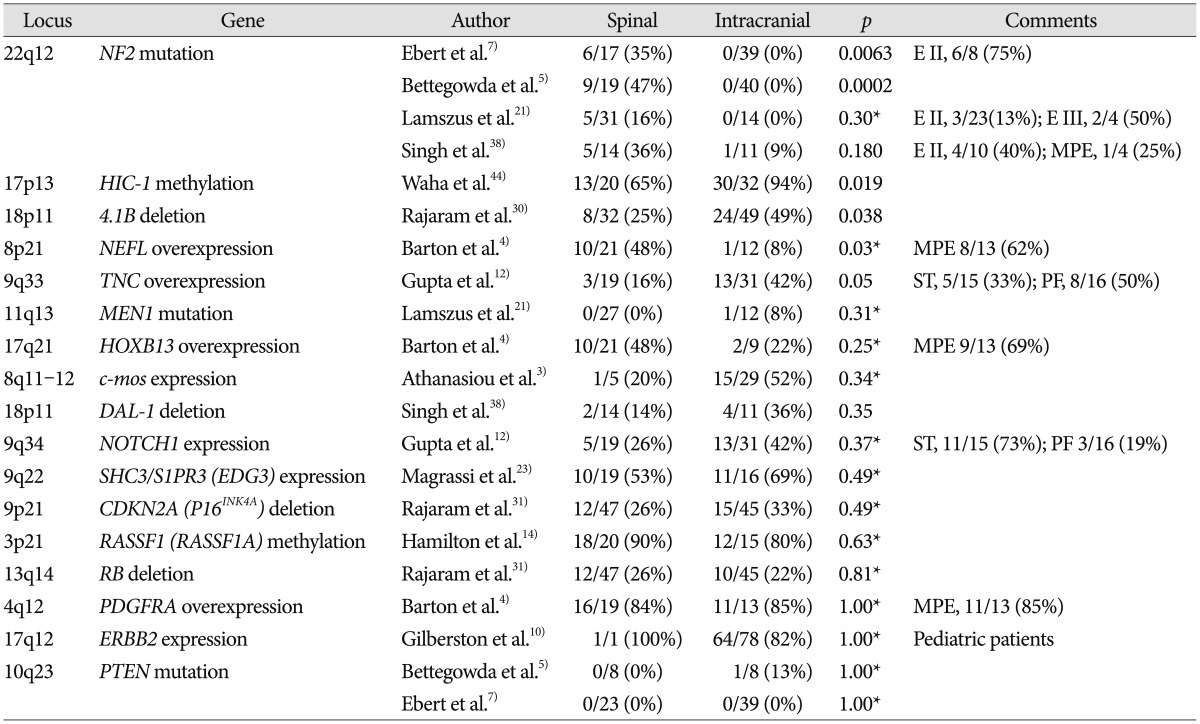
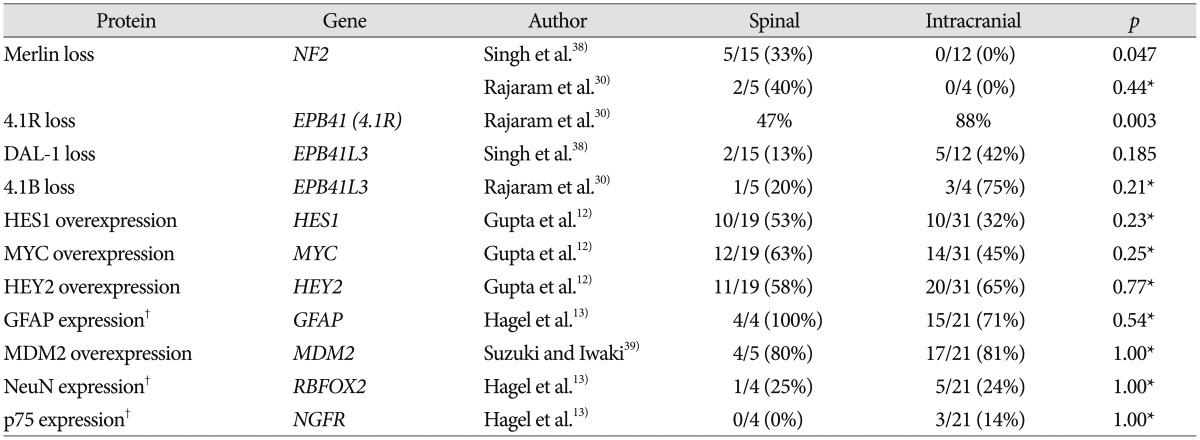
NF2 and Merlin
A variety of numerical and structural chromosomal abnormalities have been found to be associated with ependymomas; inactivation of NF2 gene, as well as sporadic mutations in NF2, on chromosome 22q12 have been well documented in ependymomas7273438). The importance of the NF2 gene to ependymoma pathogenesis is further emphasized by the observation that NF2 gene mutations and the loss of Merlin, the protein encoded by the NF2 gene, are found in 30–71% of sporadic (non-syndromal) ependymomas 938). Among 4 studies included in this review, 2 studies showed that mutation of NF2 gene is observed only in spinal ependymoma and not in intracranial ependymoma, as shown in Table 157). Another study reported that NF2 loss was strongly associated with spinal ependymoma at the protein level, Merlin, whereas at the DNA level, this was only a trend38). Some studies reported that NF2 mutations were found in a high percentage of spinal ependymoma cases (WHO grade II) especially and in few cases of MPE (WHO grade I), subependymoma (WHO grade I), and anaplastic ependymoma (WHO grade III) as well as all intracranial ependymomas727).
HIC1
Hypermethylated in cancer 1 (HIC1) gene is located on chromosome 17p13.3. Although only one study dealt with HIC1 gene in ependymoma, it clearly demonstrated that low or absent expression of the HIC1 gene is frequently found in human ependymomas and that hypermethylation was significantly more common in intracranial ependymomas than spinal tumors, as shown in Table 1 (intracranial : 94% vs. spinal : 65%; p=0.019)44). A chromosomal study reported that loss of chromosome arm 17p DNA sequences was common in sporadic pediatric intracranial ependymomas and many different human tumors, including gliomas and medulloblastomas43).
4.1B, DAL-1, and 4.1R
Both 4.1B and DAL-1 genes are located on chromosome 18p11 and are involved in different mechanisms that modulate cell growth, motility, adhesion, and cytoskeleton organization45). Inactivation of 4.1B and DAL-1 gene expression may lead to tumorigenesis and/or promote tumor progression45). One study reported that 4.1B deletions were commonly observed in intracranial ependymoma compared with its spinal counterpart in Table 1 (intracranial : 49% vs. spinal : 25%; p=0.038), and protein 4.1B loss was seen in 3 of 4 intracranial tumors and 1 of 5 spinal cord tumors in Table 230). Losses of DAL-1 gene and DAL-1 protein were more frequently observed in intracranial ependymomas than in the spinal cord form, although this difference did not reach statistical significance38). Among another proteins in the 4.1 family in Table 2, loss of protein 4.1R expression was statistically associated with intracranial location (intracranial : 88% vs. spinal : 47%; p=0.003) and young age (pediatric : 93% vs. adult : 48%; p<0.001), although presence of 4.1R deletion did not differ substantially between subsets according to tumor location and age30).
NEFL, HOXB13, and PDGFRA
MPE is regarded to be distinct molecularly from intracranial ependymoma as well as other spinal ependymomas. MPE is characterized by high expression levels of some genes, including NEFL, HOXB13, and PDGFRA4). A previous study reported that NEFL immunoreactivity in the spinal ependymoma was substantially high compared to that in intracranial ependymoma in Table 1 (spinal : 48% vs. intracranial : 8%; p=0.03)4). NEFL immunoreactivity was positive in 8 of 13 (62%) MPE cases, which is very high compared with subependymoma and ependymomas in both spinal and intracranial region. Another study reported that high expression of NEFL could predict a longer progression-free survival in supratentorial ependymomas2).
Both HOXB13 and PDGFRA genes were investigated in one paper and there was not a significant difference in immunoreactivity by tumor location4). This study also found that PDGFRA demonstrated high sensitivity but poor specificity for ependymoma, since most intracranial ependymoma cases were positive as well. The upregulation of PDGFRA suggests that the therapeutic targeting of this receptor tyrosine kinase may be an appropriate topic for future clinical trials. Several PDGFRA inhibitors have been FDA approved, including imatinib mesylate, sorafenib, and sunitinib4).
TNC and NOTCH1
Tenascin C (TNC) and NOTCH1 genes are located on chromosomes 9q33 and 9q34, respectively, and are involved in central nervous system embryogenesis. Previous studies have reported the gain of 9q in ependymoma, which is where TNC and NOTCH1 genes are located12162336). One study analyzed the correlation between TNC and ependymoma and reported that the immunoexpression of TNC was higher in intracranial ependymoma than in its spinal counterpart (p=0.05)12). In this study, the immunoexpression of TNC was positive in 50% of posterior fossa cases compared with 19% of spinal ependymoma cases and 31% of supratentorial ependymoma cases. Immunotherapy using radiolabeled anti-TNC antibodies has shown promising results for hematological malignancies and brain tumors32). Immunotherapy using anti-TNC antibodies may be a useful in the future.
One study showed no significant difference in NOTCH1 expression between intracranial and spinal ependymomas in Table 1 (intracranial : 42% vs. spinal : 26%; p=0.37). However, NOTCH1 showed significantly higher immunoexpression in supratentorial tumors (73%) in comparison to infratentorial (19%; p=0.001) and spinal (26%; p=0.01) tumors12). Notch pathway activation leads to the overexpression of the target genes HES1, HEY2, and MYC. The expression levels of the proteins (HES1, HEY2, and MYC) of these genes were not substantially different according to tumor location in Table 212). A Notch pathway enzyme, γ-secretase inhibitors may represent a promising therapeutic option for supratentorial ependymomas in future1229).
SHC3 and S1PR3
SHC3 and S1PR3 genes are located on chromosome 9q22.1-2. Dysregulation of SHC3 expression is involved in the survival of anaplastic astrocytomas and glioblastomas22). The S1PR3 gene is also known as EDG3 and likely contributes to the regulation of angiogenesis and vascular endothelial cell function. The co-immunoprecipitation of Shc3 and EDG3 proteins was reported in ependymomas with amplification of SHC3 and EDG3 genes, which suggests that the 2 proteins co-operate and are important for ependymomas23). However, the differences in gene mutations and protein overexpression were not substantial between spinal and intracranial ependymomas.
MEN1
The MEN1 gene is located on chromosome 11q13, a region that is involved in allelic losses and rearrangements in ependymomas. Ependymomas have been described in patients with MEN1 syndrome, which is characterized by the development of multiple endocrine tumors6). However, mutations in the MEN1 gene have been described in only a small fraction of recurrent ependymomas6). In one study, only 1 intracranial ependymoma patient had an MEN1 mutation among the 12 intracranial and 27 spinal ependymoma patients21). The patient with the MEN1 mutation exhibited lesion recurrence twice and metastasis.
RB and CDKN2A (P16INK4A)
Retinoblastoma susceptibility (RB) gene on chromosome 13q14 and cyclin-dependent kinase inhibitor 2A (CDKN2A) gene, also known as P16INK4A, on chromosome 9p21 are key tumor suppressor genes in a cell cycle regulatory pathway that is commonly inactivated in a wide range of cancers. The disruption of either RB or CDKN2A gene leads to deregulated cell proliferation and supports tumor progression37). Several studies reported that there were no significant genetic associations of 9p and 13q with ependymoma grade, recurrence, or death, suggesting that 9p and 13q deletions do not have obvious associations with tumor grade, age, location, or overall prognosis in Table 11131). Therefore, they might not play a prominent role in the malignant progression of ependymomas31).
c-mos
c-mos, the proto-oncogene located on chromosome 8q11-12 in humans, encodes mos, a 39-kD protein that is a component of the mitogen-activated protein kinase transduction pathway3). In one study, almost half of the ependymal tumors were immunopositive for mos, and overexpression of mos identified a biologically aggressive subgroup of ependymal tumors3). However, only 5 spinal ependymoma cases were enrolled among the 34 tumor cases, and the expression incidence of c-mos gene did not differ significantly by location in Table 1 (intracranial : 52% vs. spinal : 20%; p=0.34).
RASSF1 (RASSF1A)
Ras association domain family protein 1, isoform A (RASSF1) gene is located on chromosome 3p21.3 and has been shown to be involved in a variety of malignancies, including brain tumors such as gliomas and medulloblastomas14). Recent evidence has also suggested that the extensive hypermethylation of tumor suppressor genes, including CDKN2A, CDKN2B, HIC1, RASSF1A, CASP8, MGMT, and TP73, is an important mechanism in the pathogenesis of ependymoma33). Rajaram et al.31) reported that extensive hypermethylation across the RASSF1 CpG island was detected frequently in 18 of 20 (90%) spinal ependymomas and 12 of 15 (80%) intracranial ependymomas. The incidence of RASSF1 hypermethylation was not different between spinal and intracranial ependymomas. Other researchers reported that both supratentorial and spinal ependymomas frequently displayed RASSF1 gene hypermethylation, whereas posterior fossa tumors did not33). Thus, the absence of RASSF1A hypermethylation may be a diagnostic indicator of posterior fossa ependymoma.
ERBB2
ERBB2 gene is a member of the RTK I family and is located on chromosome 17q12. This gene encodes a member of the epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases. The ErbB2 receptor was reported to be frequently overexpressed in childhood intracranial ependymoma10), with the expression of ErbB2 identified in 82% of tumors. However, only one spinal ependymoma case was enrolled among the 121 tumor cases, and the expression difference by tumor location was not significant.
Another study demonstrated that Merlin negatively regulated spinal neural progenitor cell survival and glial differentiation in an ErbB2-dependent manner and that NF2-associated spinal ependymomas exhibited increased ErbB2 activation, indicating that ErbB2 may be a potential therapeutic target for NF2-associated spinal ependymoma9). The importance of ERBB2 in NF2-associated tumors is also highlighted by recent research showing that lapatinib inhibits vestibular schwannoma growth1).
MDM2
Oncogene MDM2, localized on chromosome 12q14-15, encodes a nuclear-localized E3 ubiquitin ligase. The encoded protein, MDM2, can promote tumor formation by targeting tumor suppressor proteins, such as p53, for proteasomal degradation. Thus, the protein is believed to act as a cellular regulator of the p53 protein39). One previous study showed that MDM2 was overexpressed at similar levels in intracranial and spinal ependymomas in Table 2 (intracranial : 81% vs. spinal : 80%; p=1.00)39).
TP53
The TP53 tumor suppressor gene on chromosome 17p13.1 is frequently mutated in human cancers, and it is important in the pathogenesis of other central nervous system tumors8). One study found that TP53 was mutated in only 1 of 31 ependymomas patients8), and another study reported that TP53 was mutated in only 1 of 16 patients5). Therefore, TP53 does not seem to be important in the pathogenesis of ependymomas, unlike other brain tumor types in which p53 mutations play a role in the progression of tumors8).
PTEN
PTEN gene, located on chromosome 10q23, has been proposed to be a candidate tumor suppressor gene that is inactivated in multiple cancers, including glial tumors. Three studies investigate the relation between PTEN and ependymoma, and all studies found that PTEN mutations are rarely present in ependymoma5752). Only one patient with a PTEN mutation had an intracranial tumor, but this patient also had a TP53 mutation5).
GFAP, NeuN, and p75
Prior study suggested that immunohistochemical (IHC) expression of p75, NeuN and GFAP differed in ependymomas depending on tumor topography supporting the view of divergent cells of origin13). They showed that glial markers such as NeuN and GFAP were preferentially expressed in infratentorial lesions, whereas neuronal markers such as p75 were found in supratentorial tumors, which reached statistically significant difference between supratentorial and infratentorial ependymoma for p75, GFAP, and NeuN13). However, the difference between spinal and intracranial tumors did not show a statistical significance for p75, GFAP, and NeuN.
CYTOGENETIC ABERRATIONS
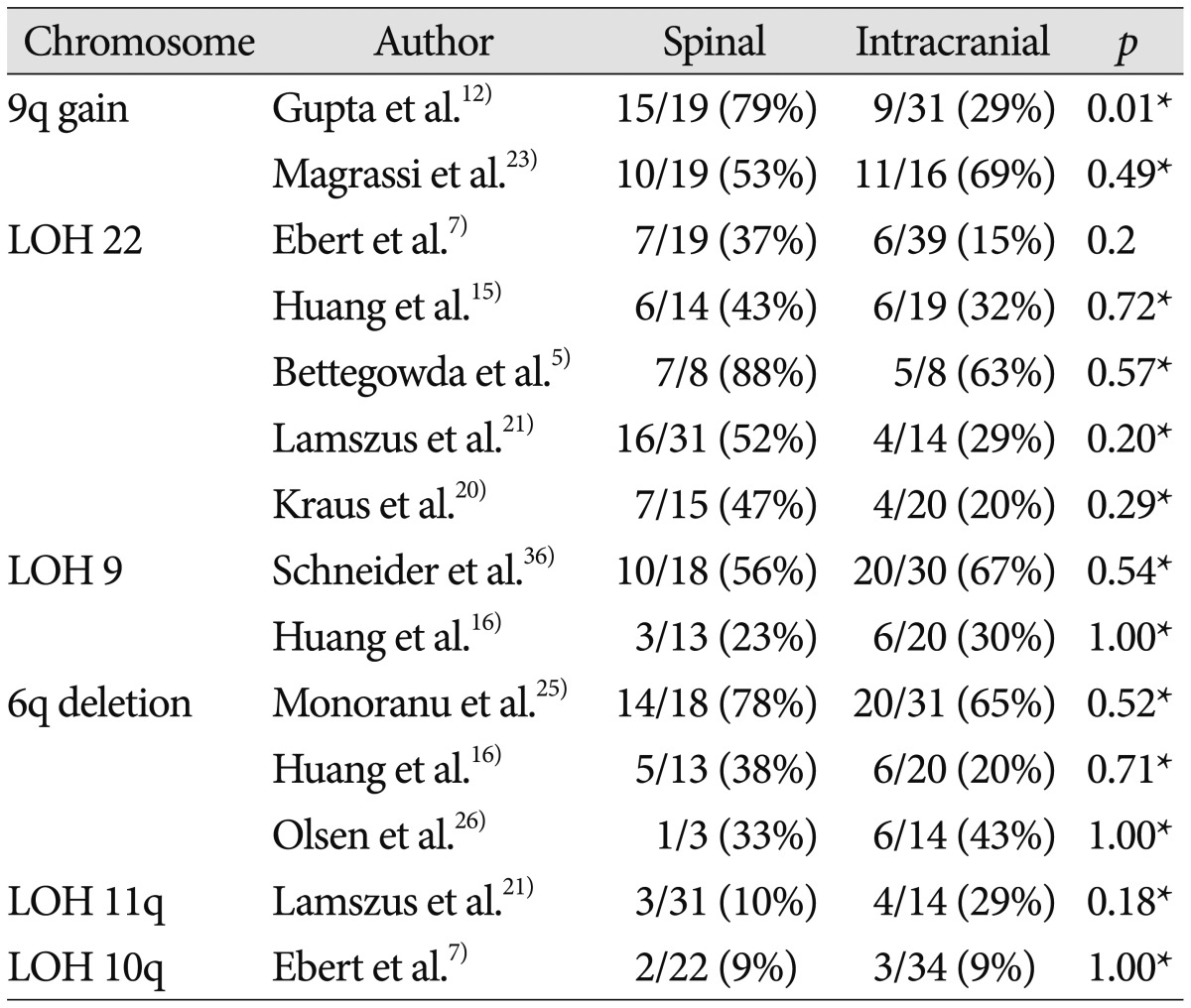
Chromosome 9q gain and loss of heterozygosity (LOH) 9
Gene amplification is an important mechanism to enhance gene expression in many tumors. Large gains and losses of the long arm of chromosome 9 have been repeatedly described in studies using cytogenetic and molecular analyses of ependymomas, and 9q gain was mapped to 9q12-32 and 9q22-31 intervals122351). A study that included 50 patients reported that chromosome 9q33-34 gain was more frequently observed in spinal ependymoma than in intracranial counterpart in Table 3 (intracranial : 29% vs. spinal : 79%; p=0.001), which showed a statistically significant difference12). Another study involving 35 patients reported that chromosome 9q22-22.2 gain was commonly detected in both spinal and intracranial ependymomas in Table 3 (intracranial : 69% vs. spinal : 53%; p=0.49)23).
Two studies investigated 9q deletion in ependymoma1636), and neither found a substantial difference between spinal and intracranial ependymomas. In a study that included 48 patients with ependymoma, 9q deletions, particularly of DCR1, were found significantly more often in supratentorial tumors than in tumors with an infratentorial location in pediatric patients (p=0.007)36).
LOH 22q
The most frequent genetic change in sporadic ependymomas is monosomy 22, suggesting the presence of an ependymoma tumor suppressor gene on chromosome 2215). The majority of relevant studies reported that LOH of 22q was more frequent in spinal ependymoma than in intracranial ependymoma, although this difference did not reach statistical significance57152021). Although NF2 gene mutations are observed distinctively in spinal ependymoma, no clear association between LOH 22 and NF2 mutations has been found. Hence, these data may either suggest the presence of another tumor suppressor gene on chromosome 22 or result from a chromosomal instability causing the random deletion of genomic material57).
Chromosome 6q deletion
Losses and rearrangements of genetic material on chromosome 6q are frequently detected in human malignancies, including central nervous system tumors such as gliomas25). Three studies reported a correlation between chromosome 6q deletion and ependymoma162526), and the incidences of 6q deletion between spinal and intracranial ependymomas were similar.
LOH of 10q and 11q
In studies of LOH of chromosomes 10 and 11, no correlation between the tumor location and the LOH was observed721). One study found that LOH of 10q was observed regardless of tumor location, showing a trend related to tumor grade (WHO grade III : 24%; WHO grade II : 4%; WHO grade I : 0%)7). Another study reported that LOH of 11q was associated with neither tumor location nor tumor grade21) and that there was a highly significant inverse association between LOH 11q and LOH 22q, which suggests that loss of genetic information on either 11q or 22q could represent independent and alternative mechanisms involved in ependymoma pathogenesis.
CONCLUSION
Spinal ependymoma has been found to be quite different from intracranial ependymoma in genetic studies, and the favorable prognosis in spinal ependymoma may be due to these genetic differences. Ependymoma in the spinal cord may be related with NF2 mutations, NEFL overexpression, and 9q gain. Its intracranial counterpart may be related with HIC-1 methylation, 4.1B deletion, and 4.1R loss. A more detailed understanding of these various genetic aberrations may enable the identification of more specific prognostic markers as well as the development of customized targeted therapies.
 XML Download
XML Download