

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Gliofibroma is an extremely rare neoplasm specified by biphasic morphology of both astrocytic and mesenchymal components. Since Friede first reported this unusual tumor in the medulla oblongata of a 3-year-old child in 1978, only about 30 cases have been reported to date8910). The tumor commonly occurs in the young, less than 20 years old. Because of the paucity of clinical and pathological information regarding this type of tumor, it has not been yet included as a distinct entity in the current World Health Organization classification of tumors of the central nervous system (CNS)912).
In this report, we describe a case of intracranial gliofibroma in the medial temporal area in a 61-year-old woman. Radiological, intraoperative, and histopathological findings of the tumor are discussed. To our knowledge, this case of intracranial gliofibroma is in the oldest patient ever reported.
CASE REPORT
A previously healthy 58-old-year woman was referred to our hospital because of abnormal findings on computed tomography (CT), which was incidentally taken for evaluation after minor head trauma. She had some minor hearing disturbance of the right ear a few years previously, but hearing was serviceable. In addition, there were no abnormal symptoms or signs, such as facial motor or sensory deficits, diplopia, seizure, or motor or sensory deficits of extremities.
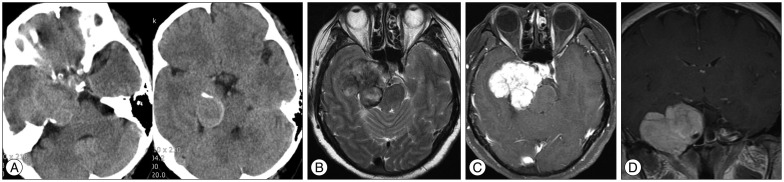
CT showed an iso-attenuated mass with a lobulating contour on the right medial temporal area. The lesion was destroying the petrous portion of the temporal bone with peripheral calcification density involving the right cavernous sinus and posterior fossa from the pons to the midbrain (Fig. 1A). Magnetic resonance (MR) imaging revealed that the tumor had iso-signal intensity on T1-weighted imaging (T1WI), heterogeneous and relatively low density on T2-weighted imaging (T2WI) (Fig. 1B). Also it was enhanced well by Gadolinium (Fig. 1C, D). The lesion caused chronic otitis media by blocking the right eustachian tube, which might have caused a hearing disturbance. On angiography, there was minimal tumor staining from a dural branch of the petrous internal carotid artery. After considering all imaging findings, our preoperative radiological diagnosis was considered to be a meningioma.
A right-sided pterional craniotomy was performed after removing a zygoma. The tumor was avascular yellowish tumor. It was too hard to be resected even using a Cavitron ultrasonic surgical aspirator. The tumor was subtotally removed except medial portion encasing the left internal carotid artery. There was no abnormality on intraoperative neuromonitoring during surgery. Immediately after surgery, she presented right peripheral-type facial palsy Grade IV according to the House-Brackmann grading system and mild hypesthesia along the dermatome of the trigeminal nerve maxillary branch.
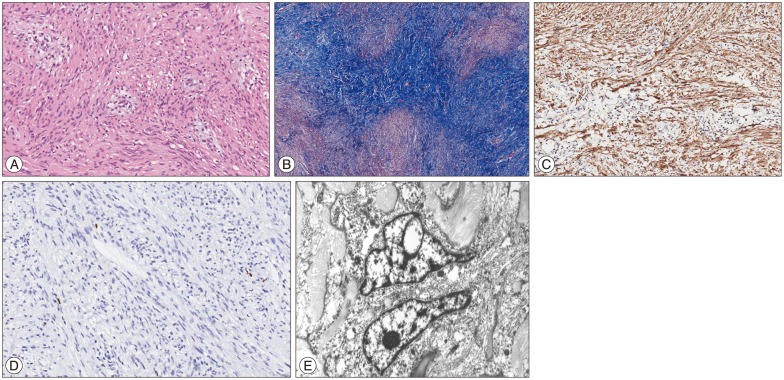
The tumor tissue was fixed in formalin and embedded in paraffin. Hematoxylin and eosin stain, the Masson trichrome stain and a few immunohistochemical stain using several monoclonal antibody were performed. Under the light microscope, the tumor was seen composed of spindle-shaped cells with prominent blood vessels and abundant intercellular collagen deposits (Fig. 2A, B). Spindle-shaped cells were presumed to be glial cells (Fig. 2C). Mitotic count was less than 1 per 10 high-power fields. There was no necrosis, prominent nucleoli, or microvascular proliferation. Immunostaining by antibody Ki-67 was almost negative, because the labeling index was below 0.5% (Fig. 2D). The other specific immunohistochemical staining was negative for epithelial membrane antigen, and positive for vimentin and S-100.
Electromicroscopy revealed individual tumor cells surrounded by abundant collagen and nuclei of tumor cells were oval to elongated (Fig. 2E). Ultimately, the tumor was diagnosed as a gliofibroma.
The patient received no additional radiotherapy or chemotherapy, and was regularly followed up until 4 years after surgery. Although she had recovered facial motor function to House-Brackmann Grade II palsy, facial hypesthesia never fully recovered. Until the last serial follow up MR, there had been no tumor growth.
DISCUSSION
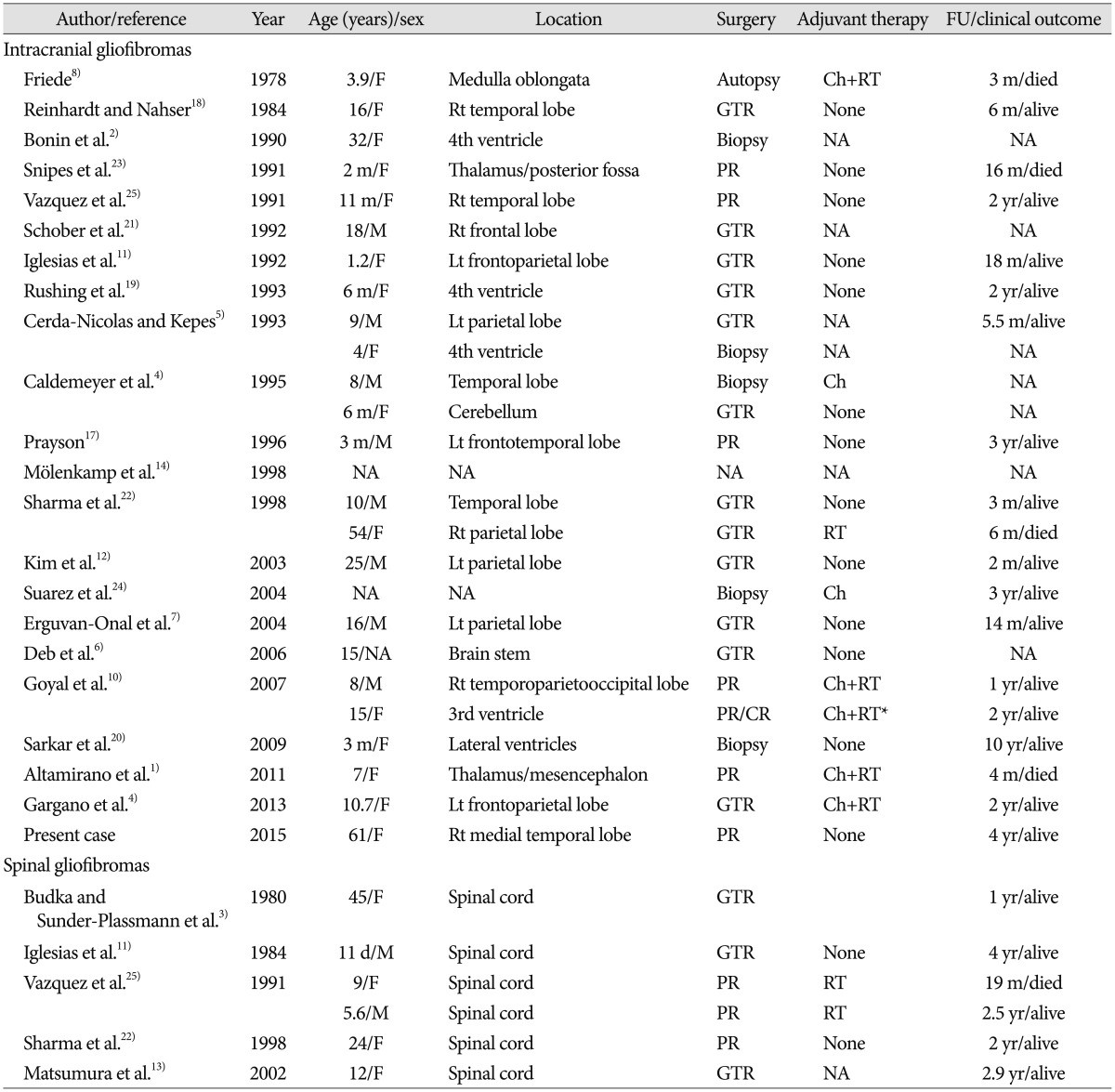
The detailed clinical information for a total of 32 patients published to date is shown in Table 1. Although there is no clear consensus about this rare tumor concerning tumorigenesis, biological behavior, and prognosis, some common characteristics in the clinical, radiological, and histopathological aspects were found.
First, gliofibromas have a tendency to affect children more than adults despite unknown predilection age6910111520). Among 32 patients, only six (19%) including our patient were over 20 years old. There were only three middle-aged patients more than 40 years old. A female predominance of 2 : 1 has been noted by analysis of previous reports46). Furthermore, the majority of gliofibromas, with the exception of five cases in the infratentorial area and seven cases in the spinal cord, had a supratentorial location, although the tumor has been described as arising in both supratentorial and infratentorial areas61012).
Second, some reports described radiological characteristics of gliofibromas, although no specific radiological features allowed a preoperative diagnosis of gliofibroma415). CT shows a well-demarcated mass with heterogeneous or homogeneous contrast enhancement. On MR images, gliofibromas appear to have various signal intensities including iso- to hyperintense on T1WI, and hypo- to hyperintense on T2WI. T2WI in our case showed an isointense mass with partially hyperintense regions. In particular, such low signal intensity on T2WI is thought to result from the reflection of the dense connective tissues as one of the peculiar characteristics in gliofibromas. Because these radiological findings include an extra-axial location, preoperative radiological misdiagnosis as a meningioma has been reported, as was the case in our patient6).
The most peculiar characteristics of gliofibromas are in the histopathological findings. Gliofibromas have peculiar 'biphasic' features composed of glial and mesenchymal components69101215172022). In our patient, the tumor clearly showed a similar biphasic appearance of mixed fibrillary astrocytes and mesenchymal components. For this reason, gliofibromas should be distinguished from other collagen-producing CNS neoplasms such as gliosarcomas and desmoplastic infantile astrocytomas or gangliomas (DIA/DIG)9101215). However, gliofibromas can be distinguished in several histopathological aspects. Although DIA/DIG have a prominent desmoplastic component similar to gliofibromas, it is usually a benign cystic tumor located on the brain surface and it has no mesenchymal component912). Moreover, gliofibroma can be distinguished from gliosarcoma by the absence of a clear-cut malignant mesenchymal component.
Although there are some exceptional cases, the majority of gliofibromas have a benign histology such as no necrosis, no microvascular proliferation, and a very low MIB-1 labeling index like this case691012151620). In addition, electron microscopic and chromosomal studies of gliofibromas have only been reported occasionally, and have not yet provided conclusive information.
Radical surgical resection as an initial management seems to be the treatment of choice. Most of the published cases with gliofibroma presented a benign tumor nature, indolent clinical course, and showed no definite evidence of disease progression. However, some cases with a poor outcome have also been reported9101624). Though some patients underwent adjuvant treatments such as radiation therapy or chemotherapy, the role of radiation therapy and chemotherapy is still controversial, and the prognosis of this rare tumor is not clearly understood. In our case, there was no disease progression during 4 years of follow-up, even if partially resected. However, long-term follow-up and more accumulated clinical experiences should be mandatory to understand this rare tumor.
CONCLUSION
In this report, we presented a case of intracranial gliofibroma in a 61-year-old woman. It is rare tumor mostly reported from pediatric patients. Also, it has characteristic biphasic microscopic morphology. We believe this case adds support to understanding more fully the nature and biological behavior of this rare tumor.
 XML Download
XML Download