

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Craniosynostosis is a congenital anomaly of cranial deformity and stenosis due to early fusion of the cranial sutures. Craniosynostosis is divided into two groups, simple or complex craniosynostosis. Simple craniosynostosis has single suture synostosis and complex one has multiple synostoses which shows more severe cranial deformity and stenosis. According to associated developmental lesions, isolated (non-syndromic) craniosynostosis shows only cranial deformities caused by synostosis and may have secondary neurologic or ophthalmologic manifestations which are derived from early fusion of cranial sutures. Syndromic craniosynostosis quite often shows multiple synostoses, associated with hypoplasia of the maxillary bone and other skeletal developmental deformities, occurring only in approximately 5–15% of all cases of craniosynostosis1040). There have been reported numerous numbers of diseases belonging to syndromic craniosynostosis. Crouzon syndrome, Apert syndrome, Pfeiffer syndrome and Saethre-Chotzen syndrome are relatively common in syndromic craniosynostosis. Cloverleaf skull should be considered as syndromic craniosynostosis, because this type of the cranial deformity is associated mostly with Pfeiffer syndrome. Characteristic manifestations of each disease, such as hypoplastic maxillary bone, fused fingers or toes, exophthalmos, broad thumbs, and fused elbow joints and so on, are important for clinical diagnosis. Most diseases in syndromic craniosynostosis are systemic due to genetic alterations, such as FGFR1, 2 or 3, TWIST and so on59). However, many reports have failed to show a close relationship between genotype and phenotype in patients with syndromic craniosynostosis, except for Apert syndrome. So far, the clinical and neuroradiological evaluation is much more important to make an individual treatment plan for each patient with syndromic craniosynostosis.
In the present report, clinical implications of treatment in considerations to the physiological changes in syndromic craniosynostosis are reviewed with an especial focus on the neurosurgical aspects.
Go to : 
LESIONS ASSOCIATED WITH HYPOPLASTIC MAXILLARY BONE
Upper air way obstruction due to hypoplastic maxillary bone
Initial thread to death early after birth in syndromic craniosynostosis is not increased intracranial pressure (ICP) but upper air way obstruction caused by hypoplastic maxillary bone. Paradoxical breathing, retractive breathing, no air flow through the external nostril, frequent interruption of swallowing the sucked milk are indicative of upper air way obstruction. Air way obstruction at birth is usually getting worse as the patient grows, because maxillary bone does not grow well as the oxygen consumption significantly increases with body growth. Severe air way obstruction may interfere continuous action of swallowing of sucked milk, resulting in insufficient weight gain due to poor nutrition. Alternative way of feeding should be considered, such as tube feeding. Polysomnographic examination is effective for evaluation of sleep apnea4). Upper air way obstruction should be continued to observe even after discharge from the hospital. In fact, 7% of patients with Pfeiffer syndromic died at home19) and the most possible cause of it could be air way trouble.
Tracheostomy is required in 33% of syndromic craniosynostosis patients before one year of age21). Tracheostomy should be considered even in infants showing sever stridor, apnea due to upper air way obstruction or SOa2 below 90%, although in the management of children with tracheostomy meticulous care and intensive support from patient's family are needed every minute. Maxillary bone advancement should be awaited until patient age becomes older than 7 years, because such surgical intervention in younger age may often result in redo the operation due to retraction of the advanced maxillary bone54). After maxillary bone advancement tracheostomy can be successfully removed in 43–67% of patients365). About 10% of syndromic craniosynostosis patients have lesions in tracheobronchial system, distal to the site of tracheostomy, which can't be effectively treated even by tracheostomy21).
Ophthalmological lesions
In infants or younger children whose cranial bone can be easily separated at the cranial sutures by increased ICP, chocked disc (papilledema) is not a typical finding of increased ICP. However, multiple synostoses block such a compensatory mechanism against increased ICP. Long standing chocked discs due to persistently increased ICP may result in visual loss. Chocked disc was found in 60% of children with Crouzon syndrome, and optic atrophy was in 20%29). So that optic fundi should be examined by ophthalmologists at the first examination and should be repeatedly examined afterward.
Ocular abnormalities, such as exophthalmos, strabismus, ptosis and so on, are often observed in syndromic craniosynostosis33). Severe exophthalmos may cause corneal ulcer, which might result in visual loss. Tarsorrhaphy is to be considered even in infants. Eyeball might be dislocated from the markedly small orbit, especially in cloverleaf skull, if the posterior wall of the orbit is so hypoplastic that increased ICP can easily push the orbital content anteriorly. Eyeball dislocation should be restored in a gentle manual manner as soon as possible. For the treatment of these lesions derived from hypoplastic orbit, fronto-orbital advancement (FOA) can expand the upper part of orbital cavity and the lower part of orbit can be expanded by maxillary bone advancement.
Hearing disturbance
Hypoplastic maxillary bone quite often causes auditory tube stenosis, resulting in hearing disturbance due to repeated otitis media with effusion33).
Go to :
SMALL AND DEFORMED CRANIAL VAULT DUE TO MULTIPLE SYNOSTOSES
Mechanisms of increased ICP in syndromic craniosynostosis
Small cranial volume due to multiple craniosynostosis is not the only mechanism of increased ICP in syndromic craniosynostosis. In fact, intracranial volume measured by neuroradiological methods is not small in all syndromic craniosynostosis children1449). Other possible mechanisms of increased ICP are associated hydrocephalus and obliteration of venous return from the intracranial space to the extracranial one caused by obstructed venous sinus associated with lambdoid synostosis56). Abnormally high PaCO2 by upper air way obstruction due to maxillary hypoplasia also raises the ICP. Small posterior fossa resulted from lambdoid synostosis might have some effect on the development of hydrocephalus by disturbing the movement of the cerebrospinal fluid (CSF) in severely deformed posterior cranial fossa. Also chronic tonsillar herniation caused by overcrowding in the small posterior fossa might induce to increase the ICP by obliteration of the CSF movement in the posterior fossa and between the intracranial and the spinal subarachnoid space. Among these lesions causing raised ICP, small and deformed cranial vault, hydrocephalus, upper air way obstruction and chronic tonsillar herniation can be surgically treatable.
Indications and timing of surgical treatments for cranial vault
When and how to treat children with syndromic craniosynostosis should be judged from the severities of increased ICP and of cranial deformity. Syndromic craniosynostosis shows progressive deformity of the cranial vault as growth of child. Therefore, once children show symptoms and/or signs derived from increased ICP, any decompressive procedure for small and deformed cranial vault should be promptly performed even in neonatal period. When no apparent symptoms and signs of increased ICP are observed at initial diagnosis, children are to be carefully followed up until adulthood. Prophylactic expansion of progressively deformed cranial cavity after infancy could be acceptable before the onset of apparent clinical manifestations of increased ICP or of marked cranial deformity. Recently, a retrospective study on the treatment results of syndromic craniosynostosis has been reported that 6 to 9 months of age is the ideal operative window for expansive cranioplasty, considering reoperation rates after initial expansive cranioplasty60).
Clinical evaluation of increased ICP
Children with increased ICP due to small and deformed cranial vault associated with syndromic craniosynostosis often fail to show typical symptoms such as headache or repeated vomiting. Head circumference is not always reliable for estimation of the cranial volume, because this two dimensional measurement poorly demonstrates the degree of cranial stenosis. Instead, condition of the fontanelle is quite important indicator for estimating increased ICP in infants. Apert syndrome usually shows a persistently large fontanelle with a wide separation of metopic and sagittal sutures. Fontanelle, however, is often closed during early infancy in bicoronal synostoses, such as Crouzon syndrome. Choked disc is a characteristic finding for long standing increased ICP even during infancy29), although absence of chocked discs does not exclude the presence of increased ICP.
Neuroradiological evaluation is also quite helpful for the estimation of persistently increased ICP. Plain radiographs of the cranium can be often indicative for the condition of increased ICP. Prominent digital markings are one of the most important findings of increased ICP. In healthy infants digital markings are not observed, and after one year of age they can be observed in a limited area, such as parietal area. Digital markings additionally observed in the frontal and/or occipital area or in the whole cranial vault strongly indicate a long standing condition of increase ICP. Although repeated CT should be avoided in children to minimize the amount of ionizing radiation, 3D-CT can show such findings of digital markings in the inner surface of the cranium. It can clearly show fused sutures and deformity of the skull which are helpful for surgical planning. MRI directly demonstrates no apparent conditions of increased ICP, but it can provide more precise information of brain morphology, such as size of the ventricles, tonsillar herniation, and various brain anomalies. MR venography can provide anomalous venous drainages from the intracranial to the extracranial space, which is helpful for any surgical treatment.
Patients whose evaluation by neurological or neuroradiological aspects seem to be unreliable could be candidates for direct ICP monitoring. For proper estimation of ICP, the monitoring should be continued at least 12 hours during sleep without any drugs affecting the ICP. Base line ICP over 15 mm Hg and/or minimally 3 plateaus waves with ICP more than 35 mm Hg in the duration of at least 20 minutes are regarded as increased ICP33), although there has been no consensus about the normal range of ICP in younger children.
Treatment of the cranial vault
The most possible cause of early fusions of the cranial sutures in syndromic craniosynostosis is genetic mutations, but no pharmacological target treatment has been developed yet59). So far, any surgical treatment for expansion and reshaping of the small and deformed cranial vault should be regarded as not curative but palliative one.
Various surgical treatments can be divided into 2 categories; Suturectomy (strip craniotomy) of fused sutures and expansive cranioplasty. Spontaneous reshaping after either suturectomy or cranioplasty is quite active during several months after birth but is gradually diminishing. After 12 months after birth, as the cranial bone becomes thicker, remarkable spontaneous reshaping cannot be expected15).
Suturectomy has been regarded as most beneficial for younger infants (before 3–6 months after birth) with sagittal suture synostosis. In such younger patients the cranium is so thin that bone flaps cannot be rigidly fixed and only suturectomy can be applicable. Fortunately, removal of the fused sutures induces spontaneous expanding of the small cranial vault and reshaping of the deformed cranial vault. Even in multiple suture synostoses, radical suturectomy can be successfully performed, as an initial decompression in neonates or younger infants (before 3 months after birth) with severe cranial stenosis and deformity, such as cloverleaf skull15). However, the bone defects after suturectomy tend to be ossified and fused in several months after suturectomy, because the pathological periosteum (the outer layer of the dura) promptly induces local ossification at the site of suturectomy. So that an additional surgical treatment will be required for expanding and reshaping the underdeveloped and deformed cranial vault.
Expansive cranioplasty is more radical and more effective for small and deformed cranial vault than suturectomy. Its purpose is to expand the cranial vault and also to improve the cranial deformity. Overcorrection of the small and deformed cranial vault is believed to be necessary1834), because the advanced bone flap is retracted in a late period after surgery and, if the retraction of the bone flap occurs significantly, reoperation after expansive cranioplasty is required. Advanced bone flaps can be fixed to the adjacent bones by either plates, or even no fixation (floating method; the bone flaps are not fixed). Although plates can provide most rigid fixation among these three methods, plates are found not to be enough for avoiding retraction of the bone flap especially in infants. Here is another way of expansive cranioplasty by distraction osteogenesis (DO). In DO the advanced bone flap is fixed by newly formed bones. Continuous stretching stress on the periosteum caused by distraction procedure actively products various kinds of cytokines for inducing rapid local ossification38). As expansive cranioplasty, FOA is applied for anterior cranial deformity and posterior cranial vault expansion is applied for posterior cranial deformity.
FOA by conventional method
In syndromic craniosynostosis, the most frequently affected suture is coronal suture and its most frequent cranial deformity is small anterior part of the cranial vault due to bicoronal synostoses, such as brachycephaly or oxycephaly. FOA is the most popular surgical procedure for such deformity. As the coronal suture runs down to the skull base, its premature closure induces deformity of the skull base as well as the cranial vault. In patients with bicoronal synostoses, supra-orbital portion of the anterior cranial base in addition to the frontal bone should be advanced anteriorly to expand the anterior part of the cranium3257). Two bone flaps are obtained by frontal craniotomy and osteotomy of the supraorbital portion combined with bilateral temporal bony wings. The frontal bone and the supraorbital bar are so reshaped as to fit the optimal shape of the fronto-orbital area. Then, the supraorbital bar and the frontal bone flap are advanced forward and are fixed using plates and bone pieces obtained from the adjacent bone by craniotomy. As rigid fixation of the bone flaps, absorbable plates should be used for infants and younger children. Metal (titanium) plates are not recommended for younger children because they tend to immigrate into the intradural space or to penetrate the overlying thin skin. Spontaneous reshaping of the cranial vault can be expected after surgery. However, the skin flap is not so much extended that the frontal advancement of these two bone flaps is limited, mostly up to 15 mm from their original portion at the midline.
The drawbacks of FOA by the conventional method late after surgery are significant retraction of the advanced frontal bone flap with the supraorbital bar, persistent large bony defects and depression at the pterional areas in both sides. Reoperation of expansive cranioplasty after conventional FOA is necessary in 30–100% of patients, when the initial FOA has been performed before 6 months or one year of age in non-syndromic multiple craniosynostosis32346166). Seruya et al.48) reported that major reoperation rates for bicoronal synostoses which were initially treated by cranial expansion by conventional method were more than 40% and that patients who underwent initial cranial expansion before 6 months of age showed a significantly higher reoperation rate (18.2%), compared with the rate (5%) in those who underwent after 6 months of age. de Jong et al.12) described that second intracranial intervention was needed in 14% of patients with Apert syndrome, 22% with Crouzon/Pfeiffer syndrome and 15% with Saethre-Chotzen syndrome.
Reoperation for expansion should be avoided. Re-expansion of the anterior part of the cranium is much more complicated than the initial one, because of tight adhesion of the bone flap to the underlying dura, higher risk of dural laceration during these dissection procedures and more blood loss during the operation. Also significant lack of bones for reconstruction of the cranial vault may result in unsatisfactory reshaping of the cranial shape.
FOA by DO
To overcome the above mentioned drawbacks of the FOA by conventional method, FOA by DO was introduced to the field of cranial vault. As the first application of DO to cranial vault, Hirabayashi et al.24) presented a case report of bicoronal non-syndromic craniosynostosis in 1998. Several midface advancement by DO in syndromic craniosynostosis were started to be report after this. In 2002, Imai et al.25) reported application of expansive cranioplasty by DO and FOA by DO in 20 patients including 4 syndromic craniosynostosis. Fig. 1 and 2 show plain radiographs of two representative cases who successfully underwent FOA by DO in the authors' institution.
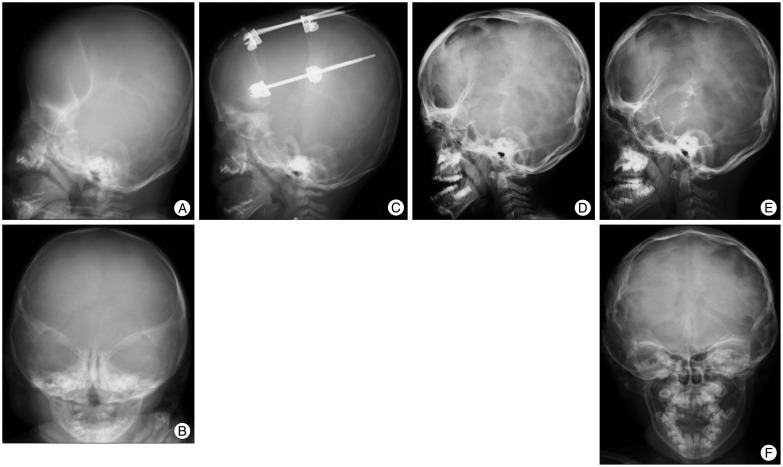 | Fig. 1Plain radiographs of a child with Apert syndrome who underwent fronto-orbital advancement (FOA) by distraction osteogenesis (DO). A and B : Before surgery (4 months old). C : At the time of completion of distraction (5 months old). D : 3 years after FOA (3 years and 8 months old). Newly developed bone was found at the bone defects. E and F : 8 years after FOA (8 years old). The advanced frontal bone with the supraorbital bar is well maintained without retraction. Maxillary bone advancement was performed later.
|
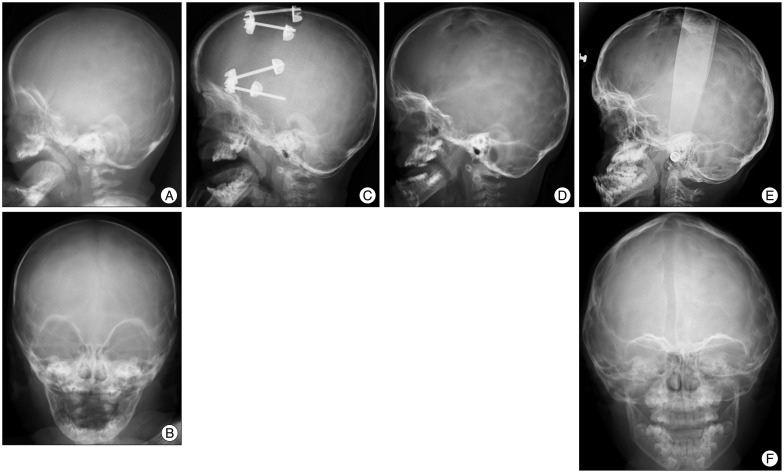 | Fig. 2Plain radiographs of a child with Crouzon syndrome who underwent FOA by DO. A and B : Before surgery (5 months old). C : At the time of completion of distraction (8 months old). The bone defect was filled by newly developed bone. D : 1 years after FOA (2 years old). E and F : 6 years after FOA (6 years old). The advanced frontal bone with the supraorbital bar is well maintained without retraction. Maxillary bone advancement was performed later. FOA : fronto-orbital advancement, DO : distraction osteogenesis.
|
In the internal distraction method2242535), as the first surgical step of FOA by DO, a single large bone flap consisted from a frontal bone and a supraorbital part of anterior cranial fossa is obtained by craniotomy for the cranial vault and osteotomy for supraorbital area, without dissecting this bone flap from the underlying dura to keep the blood supply to the bone flap. The devices are installed into the bony gaps of craniotomy and osteotomy, to distract the large bone flap to certain vectors controlled by expansion rate of each device installed at different craniotomy sites. About one week after installation of the devices, stepwise advancement by 1 mm/day is started. The skin over the bone flap is also gradually stretched. The bone flap with overlying skin flap is successfully advanced by more than 20 mm or 25 mm, which is much larger than FOA by conventional method. However, usually it takes several weeks until newly developed bone can hold the bone flap in an advanced position. The installed devices can be removed after consolidation. Among the drawbacks with DO, the main one is to remove the installed devices2).
Comparison between FOA by conventional method and FOA by DO
There have been several reports that application of expansive cranioplasty by DO mostly to isolated craniosynostosis can be more effective than conventional expansive cranioplasty5273563). Osteotomy at the supraorbital area is more technically complicated than that by conventional method. Such osteotomy is carried out without direct visual inspection. Dural laceration is one of the most avoidable complications in DO, because dural repair at the frontal base is difficult to be performed thorough a narrow osteotomy site. Recently, to improve such complexity with DO, transsutural distraction osteogenesis was newly developed and was successfully applied for children including syndromic craniosynostosis37). In FOA by DO, more radical advancement of bone flap and new bone formation at the bony gaps promptly induced by distraction are the two most significant advantages, compared with FOA by conventional method. Retraction of the advanced bone flap by FOA by DO is much less than that by the conventional method even in syndromic craniosynostosis (Fig. 1, 2). In addition, long follow-up results showed normal growth of the cranial volume after FOA by DO67).
There have been quite few reports focusing on the rate of re-expansion of the cranial vault after FOA by DO in syndromic craniosynostosis. Table 1 shows the re-operation rate after FOA by DO in syndromic craniosynostosis in the authors' institution. Surgical procedures of FOA by DO was reported elsewhere2567). Table 2 shows the operative results. Out of 41 cases who underwent FOA by DO before one year of age, 3 cases died late after FOA. Out of the remaining 38 case, 3 needed re-operation of cranial vault expansion : Two cases were Apert syndrome and one was cloverleaf skull. Two cases underwent FOA again and the remaining one with Apert syndrome needed posterior cranial vault expansion due to late onset of lambdoid synostosis. No cases older than one years at the time of initial FOA by DO needed reoperation. In the mean follow-up period of 7 years, re-operation rate was 5.2% in all cases and 7.5% in cases who underwent initial FOA by DO during infancy. These results suggest that rates of reoperation for re-expansion after FOA by DO should be low. From these results, cloverleaf skull with most severely deformed cranial vault has a high risk of re-expansion. However, one case with Apert syndrome needed re-expansion in spite of only slight retraction of the advanced frontal bone flap (Fig. 3). Considering that brain volume of Apert syndrome is significantly larger than that of healthy children49), Apert syndrome by itself might have a risk for re-operation even after sufficient expansion using FOA by DO. In addition, posterior cranial expansion for late onset of lambdoid synostosis is necessary even after sufficient expansion using FOA by DO.
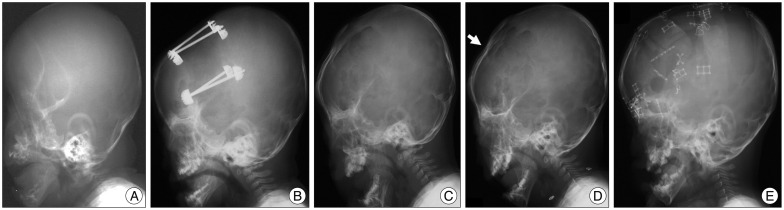 | Fig. 3Plain radiographs of a child with Apert syndrome who underwent re-expansion after FOA by DO. A : Before surgery (3 months old). B : At the time of completion of distraction (7 months old). C : 1 years after FOA (1 year 9 months old). D : At the time of diagnosis for chocked disc (2 years old). The advanced fontal bone was slightly retracted (arrow). E : 6 months after re-expansion with FOA by conventional method. Chocked disc disappeared (3 years old). FOA : fronto-orbital advancement, DO : distraction osteogenesis.
|
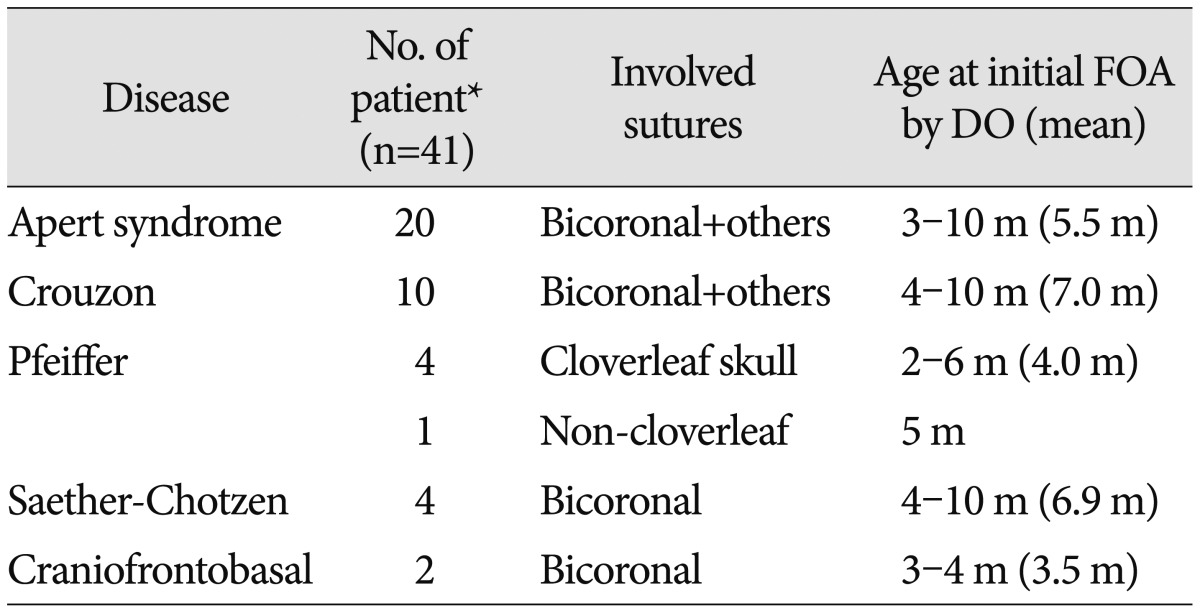
Table 1
Clinical materials of frontoorbital advancement (FOA) by distraction osteogenesis (DO) applied in the authors' institute (58 cases of syndromic craniosynostosis between 1999 and 2012)
![]()
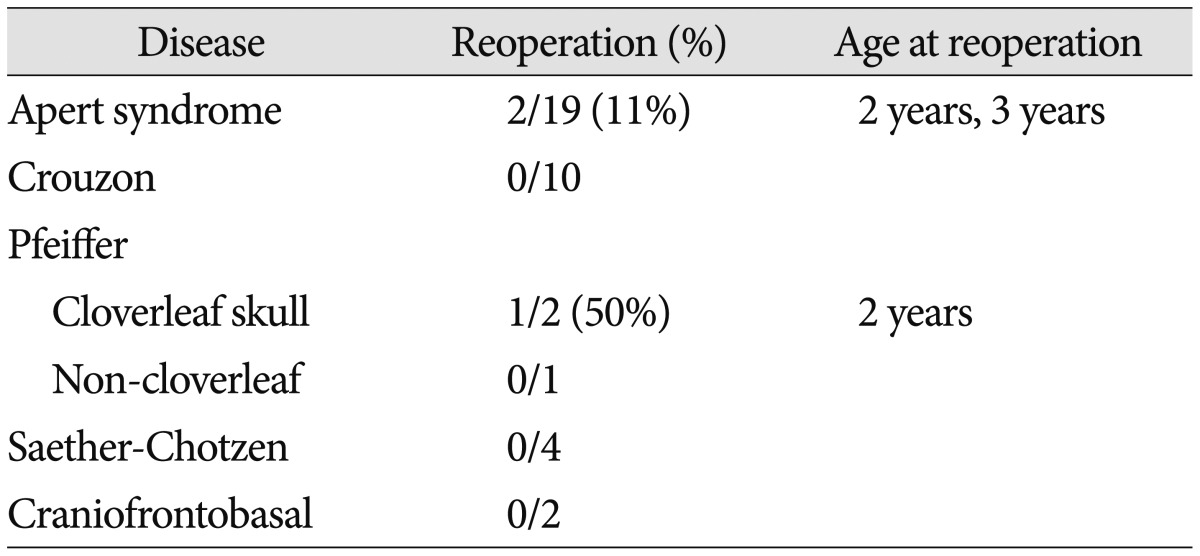
Table 2
Reoperation after frontoorbital advancement (FOA) by distraction osteogenesis (DO) applied in the authors' institute*
*The mean follow-up time of the patients was 7 years (2–15 years). Three out 41 infants at the time of surgery died in spite of successfully managing the intracranial pressure. One with Apert syndrome died from ileus at the age of one year old. Two cases with cloverleaf skull died from respiratory complications related to hypoplastic maxillary bone at the age of 1–2 years old. Reoperation was done in three patients after FOA by DO
![]()
As the most advantage of FOA by DO, sufficient expansion without significant retraction of the advanced bone flap is quite beneficial for syndromic craniosynsotosis. FOA by DO is surely considered for the expansion of anterior cranium in syndromic craniosynostosis.
Authors' proposal of timing and method for FOA in syndromic craniosynsotosis
Theoretically, FOA is be performed to expand the anterior part of the cranium at the onset of increased ICP due to multiple synostoses including cronal suture. However, FOA in early infancy could often result in re-expansion, because thin cranial bone during early infancy hardly maintain the cranial expansion. On the other hand, maximum spontaneous reshaping can be expected by cranioplasty in early infancy. So that, the authors recommend that FOA by DO is awaited until the patient age reaches 4 or 5 months when the cranial bone becomes thick enough for distraction. When early decompression for increased ICP is necessary before 4 months of age, radical suturectomy can be initially performed for spontaneous expansion of the cranial vault15) and later FOA should be planned as the second procedure. At the age of two years or older, FOA by conventional method is more beneficial than that by DO. There are two major reasons; one is that FOA by DO after infancy can induce only limited spontaneous reshaping of the cranial deformity because of the thickened cranial bone and that FOA by conventional method can provide intraoperative reshaping of the cranial deformity in addition to expansion of the anterior part of cranium. The other is that FOA by DO applied for children who can walk around freely or even run may have a persistent risk of unexpected traumatic brain or cranial injuries by the installed devices, until these devices are removed. FOA by conventional method can provide rigid fixations of the advanced bone flaps, to protect the brain from head injuries in daily activities of these children after FOA.
Posterior cranial vault expansion by conventional method
Flattening of the posterior cranial vault is caused by lambdoid synostosis and posterior part of sagittal suture synostosis42). Lambdoid synostosis is more frequent in syndromic synostosis than in non-syndromic craniosynostosis (15% in isolated craniosynostosis). Cinalli et al.7) reported more than 80% in Crouzon syndrome at 30 months of age and more than 60% in Apert syndrome at the age of 50 months.
A free-floating occipital release was reported in 1990s50), but this method often failed to maintain the posterior expansion of the cranial vault. Therefore, expansive cranioplasty by rigid fixation of the occipital bone is recommended2340). Concerning the surgical procedures, even careful dissection of tight adhesion between the occipital bone and underlying dura may retain a high risk of dural lacerations or injuries of the transvers sinuses or sagittal sinus. From these technical difficulties, posterior cranial vault expansion by craniotomy became less prevalent.
Posterior cranial vault expansion by DO
Posterior cranial vault expansion by DO is less difficult than that by FOA, because craniotomy (circumferential narrow craniectomy) without dissecting the bone flap from the underlying dura is less complicated to perform for posterior cranial vault expansion1622285262). Posterior cranial vault expansion by DO has also several advantages and disadvantages similar to FOA by DO55). Posterior cranial vault expansion by DO can be applied for infants older than 6 months. However, when the patient lies down in a supine position after the surgery, relapse may easily occur due to compression of the occipital bone.
FOA by DO versus posterior cranial vault expansion by DO
Posterior cranial vault expansion by DO has been successfully applied for syndromic craniosynostosis patients with posterior flattening due to lambdoid synostosis. Several reports have reported that, as the first step, posterior cranial vault expansion by DO is recommended to gain a more sufficient expansion of the intracranial volume2836). The effect of intracranial capacity expansion is greater by 35% in the posterior cranial vault expansion by DO, compared to that in FOA by DO, when distraction distance is almost the same between these two procedures6). A recent report, however, has described that 56% of syndromic craniosynostosis patients who underwent initial posterior cranial vault expansion by DO required frontal advancement at the mean follow-up of 4.0 years of age45). Such high reoperation rate after posterior cranial vault expansion by DO is even higher than that after FOA by conventional methods.
One of the main reasons for introduction of DO into the cranial expansion in syndromic craniosynostosis is to reduce the number of major operations by less invasive and more effective surgical procedures. Children with syndromic craniosynostosis have to undergo at least two major surgeries, cranial and midface advancement, and additionally tracheostomy, shunt surgery for hydrocephalus, followed by repeated shunt revisions or division of syndactyly in Apert syndrome. Neurosurgeons have to try to minimize the number of major operations in children with syndromic craniosynostosis. DO is quite beneficial for either FOA or posterior cranial vault expansion, compared with the conventional methods. Therefore, FOA by DO could be recommended for syndromic craniosynostosis patients with small and deformed anterior cranium due to bicoronal synostoses and/or metopic synostosis. Expansion of the posterior cranial vault by DO is beneficial only for patients with significant flattening of the posterior cranial vault. It can also be applied for progressive flattening of the posterior cranial vault by lambdoid synostosis after FOA.
Spring-assisted cranioplasty
As an internal distraction method in DO, spring-assisted cranioplasty has been applied for FOA and posterior cranial vault expansion30). The spring consists of a metallic wire that is bent, placed mainly across an osteotomy site. The bone flap is distracted gradually as the spring straightens out. Distraction of the bone flaps stops once the force of the springs reach equilibrium with counteracting tissue forces. Compared with the standard internal distraction method in DO as mentioned above2242535), the advantage of this method is that the correction of cranial shape is based on forces persistently applied to the cranial vault, consequently avoiding distraction procedures after installation of the devices (springs). The significant disadvantage is the lack of control over distraction36). A recent report of this method performed in syndromic craniosynsotosis showed satisfactory expansion of the cranial volume58). However, longer follow-up results should be required to obtain a proper evaluation of this method applied for syndromic craniosynsotosis.
Go to :
HYDROCEPHALUS
Ventricular dilatation is quite common in syndromic craniosynostosis; 30–70% in Crouzon and Pfeiffer syndromes and 40–70% in Apert syndrome33). Large lateral ventricles without any apparent symptoms and signs of increased ICP or those without increase in size should not be regarded as hydrocephalus. Incidence of hydrocephalus during infancy or childhood is 100% in the cloverleaf skull, 30% in Crouzon syndrome, 10% in Apert syndrome8). The pathophysiology of the development of hydrocephalus in syndromic craniosynostosis remains unclear, but there are several possible hypotheses such as intracranial venous hypertension due to stenosis of the jugular veins, transverse and/or sigmoid sinuses, tight posterior fossa or chronic tonsillar herniation33). Expansion of the small and deformed cranial vault may induce hydrocephalus, because decompression makes the transmantle pressure (pressure difference between the intraventricular and subarachnoid spaces) increased, resulting in progression of hydrocephalic condition.
Ventriculo-peritoneal shunt is recommended for the treatment of hydrocephalus associated with syndromic craniosynostosis, because there is no proof of non-communicating hydrocephalus1133). Ventricular catheter is inserted from the occipital horn of the lateral ventricle in patients in which FOA will be planned later. Programmable shunt valves are effective to avoid overdrainage of the CSF through the shunt and also successfully control the ventricular size, to maintain the width of the brain mantle before and after expansion of the cranial vault. After decompression, overdrainage through shunt easily induces small lateral ventricles, consequently causing an excessive epidural or subdural space after cranial expansion by conventional methods and enlarging the subarachnoid space by DO. Such an abnormally enlarged intracranial space might turn out to become intracranial hematoma or a dead space with a high risk of bacterial infection. Endoscopic third ventriculostomy can be an optional treatment because it is effective for one third of hydrocephalic patients with syndromic craniosynostosis17).
Go to :
CHRONIC TONSILLAR HERNIATION
Lambdoid synostosis develops underdeveloped posterior cranial fossa, which can induce chronic tonsillar herniation by overcrowding in the posterior fossa. Such acquired tonsillar herniation is often observed in syndromic craniosynostosis children associated with lambdoid synostosis. Cinalli et al.7) reported chronic tonsillar herniation in 70% of Crouzon syndrome, 75% of oxycephaly, 50% of Pfeiffer syndrome, and 100% of cloverleaf skull, whereas it was observed in only 1.9% of Apert syndrome. This can be explained by the fact that Apert syndrome has much less frequently lambdoid synostosis among various diseases of syndromic craniosynostosis33).
Indication for treatment is symptomatic chronic tonsillar herniation or association of enlarging syringomyelia9). The use of multimodal polysomnography technology may improve the evaluation and management of central apnea due to chronic tonsillar herniation1). Also progressive decent of the tonsils on MRI can be an indication for decompression. Prophylactic decompression for asymptomatic but severely herniated tonsils might be justified, because lambdoid synostosis progressively induces underdevelopment of the posterior fossa43). Preoperative neuroradiological evaluations are essential. MR venography shows venous drainages through the dural sinuses and the jugular veins and also sometimes delineates abnormal collateral drainage of the intracranial venous drainage46).
Posterior fossa decompression or foramen magnum decompression is effective for chronic tonsillar herniation in syndromic craniosynostosis patients10). Posterior cranial expansion, which is decompression of only supratentrial area, could improve chronic tonsillar herniation64). Even decompression of both posterior cranial vault and posterior fossa have been reported to be effective for selected patients with lambdoid synostosis4753). However, sufficient decompression seems to be more difficult than that in Chiari type I malformation. Posterior fossa decompression in syndromic craniosynostosis is performed much younger than that in typical Chiari type I malformation, because lambdoid synostosis is progressive after birth42). So that reoperation for posterior fossa decompression could be more often required in syndromic craniosynostosis44). Also abnormal venous sinuses and drainages associated with multiple synostosis including lambdoid sutures should be considered to avoid massive bleeding at the craniotomy and dural opening for decompression, if necessary. Upper air way obstruction is another risk factor associated with operation under general anesthesia. Proper indication and timing for decompression have to be sought in consideration to the factors especially associated with syndromic craniosynostosis.
Go to :
IMPAIRED COGNITIVE FUNCTION
Factors for cognitive and/or behavioral problems in patients with syndromic craniosynostosis are 1) whether or not underwent surgery, 2) severity of associated malformations, 3) age at the time of surgery, 4) presence of absence of brain anomalies or chromosomal abnormalities, 5) gender, and 6) socio-economical status and maternal intellectual quotient (IQ)1333). Concerning cytogenetic aspects, deletion of chromosome band 7p21.1. including TWIST1 gene causes Saethre-Chotzen syndrome with learning difficulties26). Cloverleaf skull associated with Pfeiffer syndrome seems to be not so well as other mild type of Pfeiffer syndrome. IQ lower than 70 is 58% in patients with Apert syndrome and 16% in those with Crouzon syndrome20). Behavioral and emotional functioning in Apert syndrome is not as good as that in Crouzon syndrome1231). Although these significant differences in cognitive functions among the diseases of syndromic craniosynostosis seem to be deeply related to the genetic alterations of each disease, aggressive various treatments are mandatory to avoid secondary insults to the brain especially due to persistently increased ICP39).
Go to :
CONCLUSION
In the multidisciplinary management of this systemic disease, neurosurgeons should become familiar with each treatment of the cranial and brain lesions and also multiple skeletal lesions, to improve the outcome. Sufficient expansion of the cranium, either its anterior or posterior part, could be obtained by DO, consequently reducing multiple major surgeries in children with syndromic craniosynostosis.
Go to :
 XML Download
XML Download