

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Spinal cord injury (SCI) can occur as a consequence of various injuries such as car accidents, falls or gunshots. Traumatic SCI represents a serious medical and scientific challenge, being the cause of permanent neurological deficits and disabilities4348). Damage to the spinal cord is not only limited to the initial injury but may also continue with the secondary damage. At the present day, it appears to be impossible to treat primary damage leading to irreversible injury. After centuries of observing the catastrophic consequences of SCI, the conception of secondary damage defined by Allen2) was about to give hope for newly developed medical solutions.
It is well known that cells can die in two mechanisms : necrosis and apoptosis. Primary injury caused by mechanical tissue damage results in necrotic cell death whereas secondary injury is the termination of a cascade of events triggered by activation of cell death mechanisms12). On this account, most of the experimental and clinical studies have focused on preventing secondary damage. Secondary injury process include edema, hemorrhage, inflammatory reaction, ischemia, the disruption of energy metabolism353). Increased glutamate levels, excitotoxicity, oxidative stres, ischemia, Ca(2+)-dependent production of nitric oxide, damage to cell membrane caused by free radicals and lipid peroxidation are some of the major components in secondary injury process56). Apoptosis is an actively regulated response that occurs after various cells have been subjected to external or internal stimuli. For quite some time, it has been well demonstrated that spinal cord lesions are greatly exacerbated by apoptosis as a result of secondary damage1224313652).
Early experiments have showed that mitochondria plays a pivotal role in SCI through apoptosis11). Mitochondria is the check point of the pathways like death receptors, granzymes, genomic damage etc. which play role in apoptosis34). Cell death as a consequence of damage has been shown to involve bax translocation to and cytochrome-c (cyt-c) release from the mitochondria, followed by caspase-3 and subsequently caspase-9 activation2731). This event is characterized by fragmentation of DNA and nuclear morphological changes36). Cyt-c, a component of the electron transport system, has been demonstrated to be essential in both intact cell and cell-free systems of apoptosis3242). The increase in intracellular oxygen radicals, decrease in ATP/ADP and NADPH levels, transcription and translation errors of Bcl-2 and damage of DNA breakdown the permeability of the mitochondria membrane. The reduction in electrochemical gradient of mitochondria transmembrane potential (Δψm) gives rise to the membrane permeability. Thus, damage to mitochondria activates different signaling pathways and cyt-c and apoptosis inducing factor (AIF) are released from the inner membrane of mitochondria and participate in effector phase of apoptosis. Changes in permeability triggered by calcium are observed in cellular death. These changes exacerbate osmotic swelling and mitochondrial lysis by reducing mitochondrial membrane potential (MMP)2837).
Settaf et al.45) have demostrated that a novel derivative of the anti-ischemic agent trimetazidine, S-15176 [N-{(3,5-di-tert-butyl-4-hydroxy-1-thiophenyl)}-3-propyl-N'-(2,3,4-trimethoxybenzyl) piperazine], was able to limit the hepatic injury induced by ischemia-reperfusion. This protective effect has been associated with the preservation of mitochondrial function and seemed to be related to the inhibition of mitochondrial permeability transition pore (mPTP) opening16) S-15176 was able to inhibit ATP synthase activity and to stimulate the hydrolytic activity of the enzyme without relation to its permeability transition pore (PTP) inhibiting property. On this account it has been proposed that S-15176 interacts with several targets in mitochondria38). S-15176 was suggested to prevent both mitochondrial swelling and cyt-c release and maintain MMP41).
In the present study, we analyzed the neuroprotective and antiapoptotic effects of S-15176 for the treatment of experimental SCI in a rat model. To our knowledge, up-to-date no reports have been recorded for the therapeutic activitiy of S-15176 in traumatic SCI.
MATERIALS AND METHODS
All experiments were conducted according to the NIH Guide for the Care and Use of Laboratory Animals after approval by the Ethics Committee for Animal Experiments of Istanbul University.
Anesthesia and animal preparation
Thirty adult male Wistar type albino rats were used. They were fed a normal diet throughout the study period and weighed 220-340 g. All the rats were starved for 24 hours before the operation. Induction of anesthesia was made by the intramuscular application of 60 mg/kg ketamine (Ketalar, Parke-Davis. Eczacıbası, Istanbul, Turkey) and 9 mg/kg Xylazine (Rompun, Bayer, Istanbul, Turkey). When necessary, 20% of the initial dose was repeated intermittently.
Surgical procedure
Dorsal regions of the animals were shaved and cleaned with antiseptic solution (Poviiodeks, KIM-PA, Istanbul, Turkey) after the anesthesia. In prone position a 25 to 30 mm midline insicion was made using interscapular distance as a reference point. T6, T7, and T8 laminae were exposed by blunt dissection and separation of paravertebral muscles laterally. Total laminectomies were performed at three levels with a micro ronguer under a microscope (Karl Zeiss OPMI Vario S88, Oberkochen, Germany). Dura mater was kept intact through the procedure. Body temperature was monitored throughout the operation and maintained at 37℃ with heating pads until the end of the anesthesia. Through further steps different procedures were applied to each group.
Spinal cord injury
SCI was performed via the drop-weight technique developed by Allen2). Laminectomies were applied to set front the duramater. Afterwards, a 5 mm diameter cylindrical glass tube was positioned at a 90° angle to the dorsal surface of the dura mater. A 4-g weight was dropped from a height of 10 cm through the tube onto the exposed spinal cord.
Experimental protocol
Animals were randomly divided into 3 groups of 10.
Group I (n=10, sham-operated control group)
In this group, total laminectomies were applied at thoracic 6-8 levels, and then the wound was closed. On the third day after surgery, five animals were perfused with 10% formaldehyde transcardially. After the rats were killed, the surgical zone was opened again and tissues were collected under microscope. On the seventh day, the same procedure was followed in the remaining 5 rats after the evaluation of neurologic status.
Group II (n=10, trauma control group)
Laminectomies were applied to the animals as described in first group and SCI was produced in the aforementioned manner. After trauma, rats were treated with Dimethylsulfoxide (DMSO) intra-peritoneally in an equivalent dose to that of S-15176. Exactly as described for Group I, the functional recovery of the animals were assesed, spinal cord samples were collected and processed at the third and seventh days postinjury.
Group III (n=10, S-15176 treatment group)
The same surgical and trauma procedures in Group II were performed to each rat in this group. S-15176 (Sigma; Farmakim, Istanbul, Turkey) was dissolved in DMSO, and administered (10 mg/kg, ip) just after weight drop trauma. Tissue collection and functional assessment are described above.
The animals were protected in a temperature-controlled room (26-28℃) for 3 days, one per cage. Bladders were manually expressed three times per day. All animals were killed by decapitation 72 h after SCI.
Light microscopy-histopathology
Tissue samples were divided into three portions and each one-third part was used for light microscopic evaluation, immunohistochemistry and TUNEL staining. Spinal cord tissues obtained at 3rd and 7th days postinjury were defined by 10% neutral formalin solution and each 2-cm cord segment was embedded in parafin 5 µm thick coronal sections were taken and put on microscope slides covered with poly-l-lysine (PLL, Sigma P-8920, St. Louis, MO, USA). They were stained with hematoxyline and eosin (H and E), and cresyl violet (CV) dyes for histopathological evaluation. Slides were examined under the light microscope at 400× magnification.
Immunohistochemistry
Immunohistochemical analysis was performed as previously described51). Immunoperoxidase staining was performed using Histostain-Plus Bulk Kits (Life Technologies, 85-9043, GrandIceland, NY, USA), including rabbit polyclonal Bax (Delta Biolabs. DB005, Muraoka Drive Gilroy, CA, USA; 1 : 50 dilution), goat polyclonal cytochrome-c (Santa Cruz Biotechnology, sc-8385, Santa Cruz, CA, USA; 1 : 50 dilution) antibodies and rabbit polyclonal active caspase-9 antibodies (Novus Bio., NB100-56118, Littelon, USA; 1 : 500 dilution). Immunostaining procedures were carried out following the manufacturer's instructions. To determine the specificity of immunostaining, sections were incubated following the above procedure, except for omission of incubation with the primary antibody. Instead of primary antibody, control serum was used as negative control.
Semiquantitation of immunoperoxidase staining
Immunostaining was evaluated using a Leica DM2500 light microscope (Leica Microsystems, Wetzlar, Germany). Bax, cytochrome-c and caspase-9 immunostainings were analyzed in the immunoperoxidase stained cord tissues in randomly selected areas. The blind and randomized mode of analysis was performed at ×400 magnification for all immunoperoxidase stained sections in per cord section. All animals were included in the data analysis.
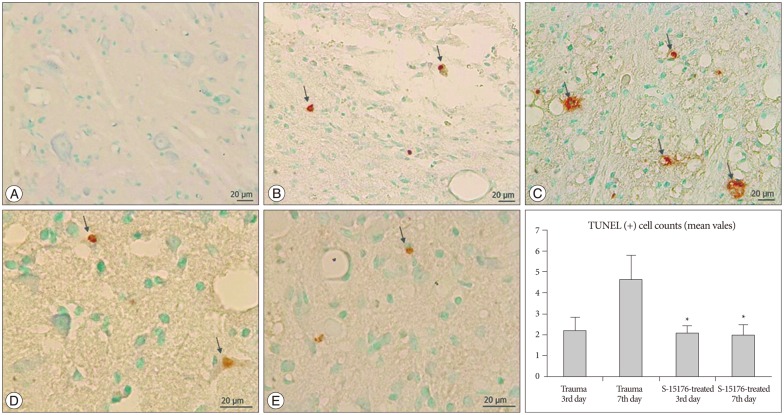
The TUNEL staining and quantification of apoptotic cells
TUNEL method was used to detect apoptotic cells in spinal cord samples that were collected at 3rd and 7th days postsurgery/injury. The in situ DNA end making method was used and it was conducted according to the protocol provided in the kit (ApopTag Plus, In Situ Apoptosis Detection Peroxidase kit, S7101-KIT, Chemicon). TUNEL analysis was performed as previously described52). For staining specificity controls, the thymus tissue of dexametazone-treated rat was used as positive control. Distilled water was used instead of Tdt enzyme as a negative control. Cells with brown nuclear labeling were defined as TUNEL positive. Staining was evaluated using a light microscope after counterstaining with methyl green.
Apoptotic index
Marked apoptotic cells were counted under high-powerfields (×400) with a light microscope. Nuclei stained with methyl green were evaluated as healthy while cells with Brown nuclear staining were considered TUNEL positive. All TUNEL positive cells in 12 different unit areas were counted on the cross-sections by a blinded researcher. These 12 fields were randomly selected from slides without discrimination between white and gray matter. Average cell per unit area number for each set of specimens (3 days and 7 days) in each group was calculated and compared.
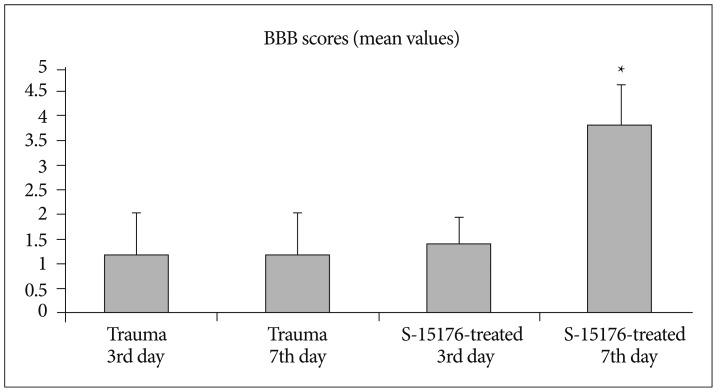
Evaluations of motor function
BBB Scale developed by Basso et al.4) is a semi-quantitative scale evaluates movement, weight support and coordination of the rats. The results were evaluated according to this scale, where 0 indicates no motor activity and 21 indicates normal performance4). Open field BBB scoring was applied to the animals at 3 days and 5 days after surgery/injury. Two researchers who were blinded to experimental protocols evaluated the functional recovery. Animals were allowed to walk freely in a 75×120 cm field where the movements of the hind limbs were closely observed and their ability to maintain postural stability was assessed with inclined-plane test.
Statistical analysis
Values were expressed as mean±SD. Statistical analysis for cytochrome-c, bax, caspase-9 immunoreaction and apoptotic cell number after the normality of variables was checked. The Kruskall-Wallis test was used and subsequently Mann-Whitney U non-parametric test was performed for significant data obtained in the Kruskall-Wallis test. p<0.05 was considered as statistically significant.
RESULTS
Histopathologic findings
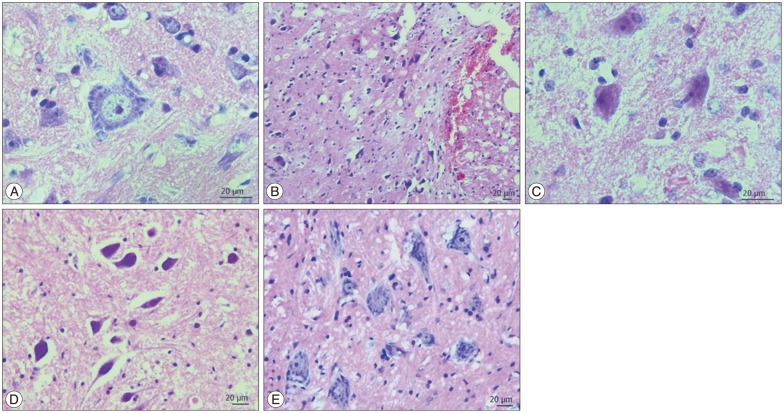
Edema, hemorrhage, necrosis and leukocyte infiltration were evaluated through histological examination. Light microscopic examination of the neural and vascular structure of cord samples revealed normal findings in control groups (Fig. 1A). Cross-sections obtained at third and the seventh days posttrauma, showed diffuse hemorrhage, cavitation, necrotic, and edematous areas especially in the gray matter. The traumatized cord segment also showed degeneration in motor neurons (Fig. 1B, C). In the specimens of the seventh day trauma group, there were hemorrhagic areas in white and gray matter, necrosis, edema and cavitation (Fig. 1C). S-15176-treated group showed more healthy cells in gray matter beside cavitation areas, hemorrhagic and edematous regions on the third day (Fig. 1D). In the seventh day of the S-15176-treated group, the cord samples revealed less cavitation and demyelinization of nerve fibers and increased number of healthy cells when compared to other groups (Fig. 1E).
Bax immunohistochemical staining
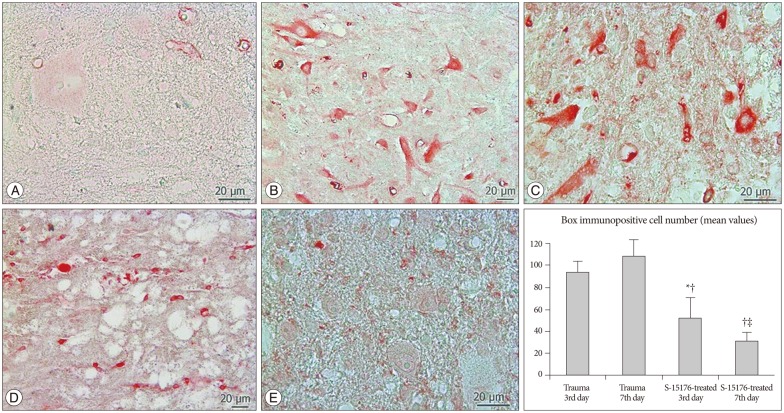
In all tissue cross-sections, Bax immunopositivity was counted by scanning of the gray and white matter. Bax immunopositivity was observed in very small proportion of cells in control group (Fig. 2A). A large number of glial cells and motor neurons have shown immunopositivity for Bax in trauma third day group (93.8±9.98) (Fig. 2B). More immunopositive cells were counted in trauma seventh day compared to the trauma third day (108.0±15.28) (Fig. 2C). Decreased Bax immunopositivity was observed in the third day of the treatment with S-15176 (52.0±19.00) (Fig. 2D). Significantly less Bax immunopositive glial cells and motor neurons were counted in the seventh day of S-15176-treatment compared to trauma groups and third day S-15176-treated group (30.8±8.33) (p<0.001) (Fig. 2E).
Cytochrome-c immunohistochemical staining
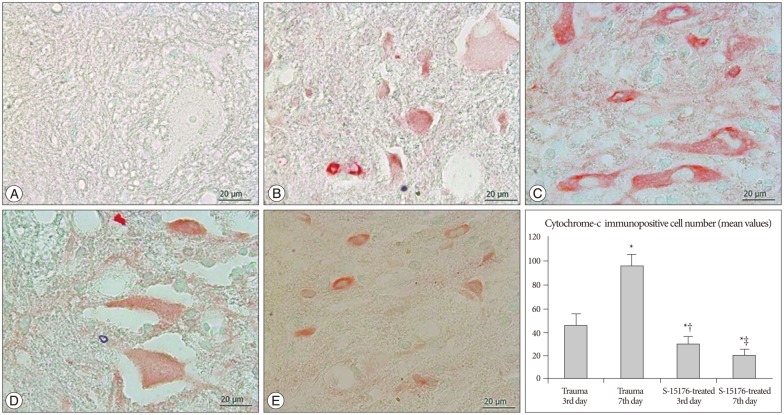
Cytoplasmic cytochrome-c immunopositivity was evaluated in all tissue cross-sections at all gray and white matter. Weak (-) cyt-c immunopositivity was observed in the control group (Fig. 3A). In trauma third day group, immune positive cells for cyt-c were significantly increased in motor neurons and glial cells (45.2±9.91) (Fig. 3B). More cyt-c immunopositivity was seen in the trauma seventh day group when compared to other groups (95.8±9.65) (p<0.001) (Fig. 3C). Decreased cyt-c immunopositivity was observed in the third day of the S-15176-treated group in all cross-section areas (29.4±6.87) (Fig. 3D). This reduction was significant statistically in the seventh day of the S-15176 treatment (20.2±5.11) (p<0.001) (Fig. 3E).
Caspase-9 immunohistochemical staining
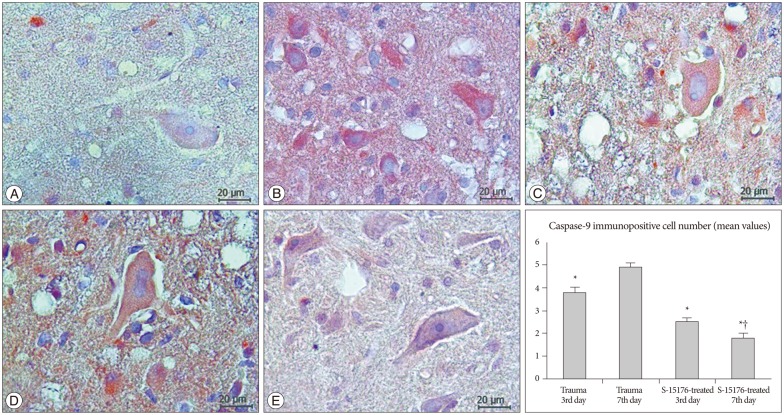
Caspase-9 immunopositivity was evaluated in all tissue cross-sections at all gray and white matter. Weak (-) caspase-9 immunopositivity was observed in the control group (Fig. 4A). Immunopositivity for caspase-9 was increased in trauma third day group compared to control group (3.80±0.21) (Fig. 4B). In the trauma seventh day group immunopositivity was increased when compared to other groups (4.92±0.16) (p<0.001) (Fig. 4C). Caspase-9 immunopositivity was significantly decreased in the third day of the S-15176-treated group in all cross-section areas (2.50±0.18) (p<0.001) (Fig. 4D). In the seventh day of the S-15176-treated group, a significant decrease in caspase-9 immunopositive cell number was observed compared to trauma seventh day and S-15176-treated third day groups (1.77±0.25) (p<0.001) (Fig. 4E).
Apoptotic cell count
Cells with brown coloured nuclei were evaluated as apoptotic in the TUNEL staining method. Apoptotic cells were counted by scanning whole gray and white matter. Apoptotic cell number per unit area was found to be (0.08±0.0) in control group (Fig. 5A). Apoptosis was commonly observed near the lesion site among mostly the oligodendrocytes and other glial cells in trauma groups (Fig. 5B, C). The number of apoptotic cells per unit was (2.20±0.62) in trauma third day group and the increase was significant when compared to control groups (p<0.05). In the trauma seventh day group, apoptotic cell number per unit area was found to be (4.63±1.16). In the S-15176 treatment groups, apoptotic cell numbers in 3rd and 7th days were significantly decreased compared to 7th days of trauma group (2.08±0.34) (1.98±0.50) (p<0.001) (Fig. 5D, E).
Results of functional neurological improvement
Evaluation of the functional recovery by open field BBB scoring revealed the scores (1.20±0.83) and (1.20±0.83) for the trauma third day group and trauma seventh day trauma group respectively. BBB score was (1.40±0.54) for the third day of S-15176-treated group and (3.80±0.83) for the seventh day af S-15176 treatment. These results indicate significant recovery for the seventh day of S-15176 treated group compared to trauma groups and third day of S-15176 treated group (p<0.05) (Fig. 6).
DISCUSSION
Interest in regeneration studies on SCI has gained momentum in the last two decades since Cheng et al.9) have demonstrated that there could be regeneration in spinal cord after injury. Moreover, experimental studies have shown that a small aliquot of axonal residue could support neurological improvement. For this reason, any attempt to increase the number of functional cells and response to stimuli must be considered as a lead for possible recovery214349).
In 1996 Katoh et al.29) have shown apoptosis following the contusion of spinal cord after trauma. Apoptosis in neurons, astrocytes, oligodendroglias and microglias of rats after SCI has been reported by Yong et al.54) and Liu et al.35) who were able to determine apoptosis in neurons and glia within 4 h after trauma. It has been demonstrated that apoptosis reaches its most intense point in neurons and glial cells on the 8th and 24th hours, respectively. It has also been stated that enlargement of the initial lesion was observed with more cell death and cavitation. Axon degeneration in oligodendrocytes in white matter on the seventh day revealed a second wave of apoptosis353646). In the trauma group of our study; edematous areas, diffuse hemorrhage, necrosis, cavitation and leukocyte infiltration were evaluated through histological examination especially in gray matter. The number of apoptotic cells had increased significantly near the lesion site among oligodendrocytes and other glial cells in trauma groups when compared to other groups. This finding has confirmed that apoptosis plays an important role in secondary injury.
Activation of mitochondria (release of cyt-c from mitochondria to cytoplasm) determines the irreversible point in apoptotic process. Mitochondria takes a critical part in the formation of cerebral metabolism, achieving homeostasis of cellular Ca2+, reactions of oxidation-reduction and orchestration of metabolic process18). Deterioration of these functions cause cell death directly or indirectly with increased cellular stress. The central nervous system trauma interrupts mitochondrial functions by impairment of cellular respiration and oxidative phosphorylation. Moreover, existing damage disrupts the intracellular Ca2+ homeostasis by inhibiting mitochondrial Ca2+ transport13). While it has been shown that noticeable changes do not exist in endoplasmic reticulum, mitochondria and Golgi bodies during the early stages of apoptosis, swelling of the outer membrane of mitochondria and release of a flavoWprotein related to cyt-c and an oxidoreductase, AIF, have been reported. Permeability changes of mitochondrial inner membrane induced by Ca2+ were observed during cell death. These changes aggravate osmotic swelling and mitochondrial lysis by reducing the mitochondrial membrane stability47).
In accordance with the findings set out above, permeability of mitochonrial membrane has become a therapeutic target. It has been shown that opening of the mPTP causes a massive swelling of mitochondria coupled with collapse of the MMP2342), total inhibition of ATP synthesis, finally leading to cell death. A growing body of evidence suggests that viability of the cell would depend on the degree of PTP opening in this process6111857). Ca2+ overload, high phosphate concentrations, ATP and ADP depletion and oxidative stress give rise to PTP opening. In isolated mitochondria, sustained mPTP opening and cyt-c release can be evoked by Ca2+ overload. A key component of the mitochondrial electron transport system, cyt-c is needed for the vitality of the cell, and as a key trigger of the mitochondrial route of apoptosis. The blockade of permeability transition prevents apoptotic neurodegeneration and, consequently, suggests a potential strategy for neuroprotection5055). This is why a group of scientists have suggested that pore opening might be a critical event in the genesis of the cell death561115). Breakdown of MMP occurs as a result of intracellular changes after SCI. Consequently different pathways of cell death are activated. PTP is also thought to be a key factor in the release of the apoptosis-inducing factors and the activation of caspases2233).
Observations on Cyclosporine A have also reinforced this hypothesis. Cyclosporine A has been proposed as the most potent inhibitor of PTP719), and it has been reported to be a protective agent which acts by inhibiting PTP2023). It has been suggested that cyclosporine shows neuroprotective effects by inhibiting mitochondrial membrane permeability, it has also been reported that it could be important for preventing cell death in experimental traumatic cord injury studies1314). Morin et al.38) have reported that search for novel PTP inhibitors should be considered with great interest, for molecules which can inhibit PTP opening can provide protection against reperfusion injury.
Trimetazidine has been shown to limit the extent of cellular injuries in experimental models of ischemia-reperfusion2644). This effect was thought to be due to the preservation of mitochondrial functions. Elimadi et al.1517) have investigated the neuroprotective effects of S-15176, Difumarate Salt which is a derivative of trimetazidine. It is thought that S-15176 inhibits mitochondrial permeability transition, prevents the early step in apoptosis by preventing collapse of the electrochemical gradient across the mitochondrial membrane. Settaf et al.45) have shown its membrane-stabilizing potential and anti-apoptotic effects on liver injury by using a rat model. It was clearly evident that S-15176 protected hepatocytes from ischemic injury and future studies would focus on mitochondrial pathways. However, it has been stated that present data did not provide detailed information on the mechanism of action. Uncertainty was resolved by a several of studies. Elimadi et al.15) have investigated the effects of S-15176 on mitochondrial functions by isolating mitochondria after ischemia reperfusion injury of liver. They have reported that S-15176 shows its effects on inner membrane and protects mitochondria by preventing passage of PTP. It has been demonstrated that S-15176 inhibits Adenine Nucleotide Translocase which regulates ADP/ATP exchange on mitochondrial membrane and is able to prevent PTP opening also in this way1). Morin et al.38) have shown that S-15176 inhibits the mtPTP induced by not only Ca(2+) and inorganic phosphate, but also by tert-butylhydroperoxide or phenylarsine oxide. In a recent study S-15176 has been proposed to be a more universal permeability transition inhibitor than Cyclosporine A or oligomycin as S-15176 was effective in suppressing the permeability transition and the subsequent cyt-c release induced by Ag(+)30).
S-15176 also inhibits ATP synthase activity but considering the protective effects of S-15176, inhibition of PTP does not seem to be related to inhibition of ATP synthase activity. An increase in transmembrane potential, ADP matrix content or phosphate uptake could be the reason for the protective effect of ATP synthase inhibition840). In an other study, Albengres et al.1) have shown that S-15176 prevents the opening of PTP totally by inhibiting adenine nucleotide translocase which regulates the exchange of ADP/ATP. Thus, Ca2+ won't be able to move out through PTP. It has been stated that S-15176 prevents the instability of the resting membrane potential and oxidation of NAD(P)H. Mitochondrial swelling has been associated with elevated levels of Ca2+ as well, that is to say protective effects of S-15176 were not only related to its antioxidant properties45). S-15176 has drawn attention with different functions in some other ways. Hamdan et al.25) have investigated the effect of S-15176 on carnitine palmitoyltransferase-1 (CPT-1) activity in rat heart and liver mitochondria. They have reported that S-15176 inhibits CPT-1 and limits the increase of fatty acid metabolism in vivo by blocking mitochondrial uptake. They have suggested that S-15176 exert its effects by switching heart and liver metabolism from fatty acid oxidation to glucose oxidation. These effects have been proposed to be beneficial in myocardial ischemia which is related with toxic metabolites caused by fatty acid metabolism.
In the S-15176-treated third and seventh day groups, the numbers of apoptotic cells per unit area were found to be reduced compared to the trauma groups. This finding indicated that apoptotic cell death was significantly prevented by the S-15176. In this study, bax, cyt-c and caspase-9 immunopositive cell numbers were increased in trauma seventh day group compared to those of the third day group. In the S-15176-treated seventh and third day trauma groups, Bax, cyt-c and caspase-9 immunopositive cell numbers were found to be significantly reduced compared to the trauma groups. These findings support the hypothesis that use of S-15176 as a Difumarate salt is able to prevent the expression of pro-apoptotic proteins, which could develop in the early stages of the mitochondrial apoptosis pathway.
In this study, after the evaluation of subject neurological functions, the average BBB values obtained from animals showed a significant increase in seventh day results of the S-15176 group compared to all the trauma groups. The locomotor abilities of the subjects of the S-15176-treated trauma groups were found to be increased compared to the subjects of the trauma groups. This finding showed that administration of S-15176 to trauma groups was able to contribute to the healing of neurological functions after SCI.
CONCLUSION
Since the first recorded use of S-15176 in an experimental model for SCI, morphological evaluation have revealed that it would prevent secondary injury in trauma groups. Here we suggest that S-15176 prevents the release of cyt-c from mitochondria by stabilizing MMP and inhibits apoptotic cell death via knockdown of caspase-9 expression. We have come to the conclusion that S-15176 should be considered as a neuroprotective agent. Therefore, future studies are highly necessary to better understand the impact of treatment strategies on outcomes in "real" patients.
 XML Download
XML Download