

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
McCune-Albright syndrome (MAS) is a rare and sporadic disease characterized by a triad of poly/monostotic fibrous dysplasia, café-au-lait macules and hyperfunctioning endocrinopathies2,9). The presence of two of the above features is sufficient to make the diagnosis. Polyostotic bone lesions and café-au-lait macules are common, while monostotic bone lesions are rare3). Endocrinopathies include sexual precocity (most commonly), hyperthyroidism, hypercortisolism, growth hormone (GH) excess and hyperprolactinemia8).
Approximately 20% of MAS patients have GH excess, which is due to a pituitary adenoma in about one-third of patients. The remaining patients have pituitary hyperplasia of GH and prolactin (PRL) secreting cells1). Relatively few adenomas from MAS patients have been studied in detail.
The presence of massive, skull-based fibrous dysplasia in some MAS patients with hypersecretory pituitary diseases often precludes the standard transsphenoidal or subfrontal approach to pituitary adenoma resection. Only a few studies have reported successful transsphenoidal approach (TSA) neurosurgical procedure and resection. Thus, this condition presents a significant therapeutic challenge for even the most experienced clinicians3,4,5).
In this case report, we report our surgical, endocrinological experience with a MAS patient with acromegaly who has been followed 3 years and adequately managed by decompression of optic nerve and near-total transsphenoidal resection.
CASE REPORT
First clinical manifestation
A 19-year-old man first visited our medical center when he was 16-years-old because of intermittent headache, dizziness and left eye visual disturbance. He had been assessed using magnetic resonance image (MRI) and electroencephalography (EEG) for these symptoms at a regional hospital. There was no abnormal finding in EEG but MR imaging showed mucoperiosteal thickening in the left maxillary sinus expanded bone in the left frontal, temporal calvarium and body of sphenoid bone with encroachment of the left optic canal. However, MR images did not show abnormal finding of pituitary gland.
On the physical examination, he had prominent left frontotemporal region and no café-au-lait macules. The results of a formal ophthalmologic consultation revealed a left and right visual acuity of 0.6 and 1.2, respectively. His visual fields and ocular fundi were normal. His height was 186 cm, which approached the 3rd percentile for that age in Korea, but there were no other definite acromegaly features. Skeletal long bone X-ray demonstrated no bony abnormality.
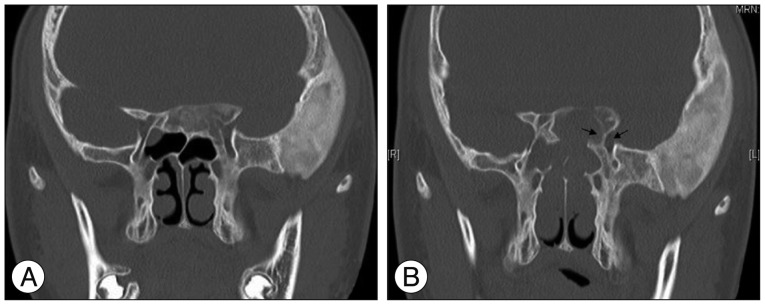
A non-contrast CT demonstrated an expanded left basiocciput and greater sphenoid wing with "ground glass" appearance, typical for fibrous dysplasia (Fig. 1A). In addition, there was left optic canal narrowing by involvement of fibrous dysplasia in CT scan. Endocrine evaluation demonstrated the following : prolactin (PRL) level 11.8 ng/mL (reference range 3.1-16.5), thyroid-stimulating hormone (TSH) level 0.75 mIU/L (reference range 0.3-6), human GH (hGH) level 3.33 ng/mL (reference range 0-4.7), cortisol level 7.2 ug/dL (reference range 5.9-26.1), ACTH level 19.6 pg/mL (reference range 0-60), luteinizing hormone (LH) level 2.1 mIU/mL (reference range 1.4-11.1) and follicle-stimulating hormone (FSH) level 3.9 mIU/mL (reference range 1.6-17.8). Bone scan demonstrated diffuse radioactive uptake in the left temporal, frontal bone, left mandible and left iliac bone that suggested active bone lesions, such as fibrous dysplasia with mild radiouptake in the left tarsal bone and left 2nd metatarsophalangeal joint.
For the prevention of visual deterioration by growing fibrous dysplasia on the skull base, he underwent an endoscopic transnasal transsphenoidal and transethmoidal approach and optic canal decompression. During the surgical procedure, a neuronavigation system (Brainlab, Feldkirchen, Germany) was used for image guidance. The middle turbinate was retracted medially to remove the uncinate process and the ethmoidal bulla was opened, allowing entrance to the anterior ethmoidal cells. Ethmoidal and sphenoidal sinus was filled with bony or cartilaginous dysplastic fibrous materials. During removal of these lesions within the nasal cavity, normal nasal anatomical structures were gradually identified with the assistance of neuronavigational guidance. The natural ostium of the sphenoid sinus was then identified and opened. Removal of intranasal dysplastic lesions provided visualization of the optic protuberance in the sphenoid roof. Bony protuberance was thickened by growing fibrous dysplasia and as a result, optic canal was narrowing. The thickened optic canal surrounded by fibrous dysplasia was carefully removed with a diamond drill. Copious irrigation with saline was important to prevent a thermal injury from the diamond drill to the optic nerve. When the optic nerve was exposed toward the optic canal, the tuberculum sellae, the thickened bony structures of the sellar floor located between the two optic protuberances, were also drilled and opened wide to relieve all areas of compression. The intracanalicular portion of the optic nerve was identified along the lateral wall of the sphenoid sinus, superior to the internal carotid artery. Postoperatively, there were no complications such as cerebrospinal fluid leakage, nasal bleeding, newly developed visual symptoms or diabetes insipidus. Postoperative follow-up CT scan revealed relief of left optic canal by removal of the fibrous lesion (Fig. 1B).
Second clinical manifestation
During the follow-up period of two years, the patient newly developed prominent supraorbital ridges, prognathism, large spade-like hands and a deep voice.
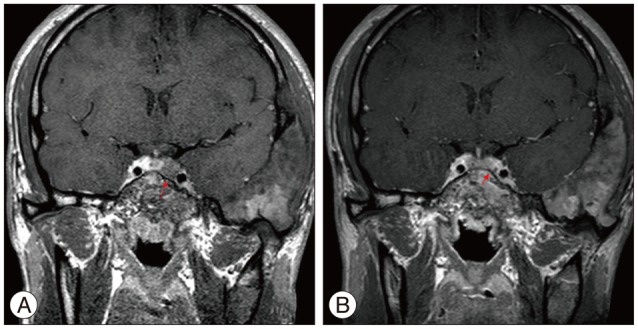
Sellar magnetic resonance (MR) images revealed slightly prominent posterior pituitary gland with newly developed mass-like lesion on the left side of pituitary gland (Fig. 2A).
Hormone evaluation revealed elevation of hGH (10.39 ng/mL) and insulin-like growth factor 1 (IGF-1) : [838.3 ng/mL (reference range 49-642)]. The other hormone levels were in the normal range : PRL 7.8 ng/mL, TSH 1.61 mIU/L, cortisol 8.6 ug/dL, ACTH 50.2 pg/mL, LH 3.01 mIU/mL and FSH 3.58 mIU/mL.
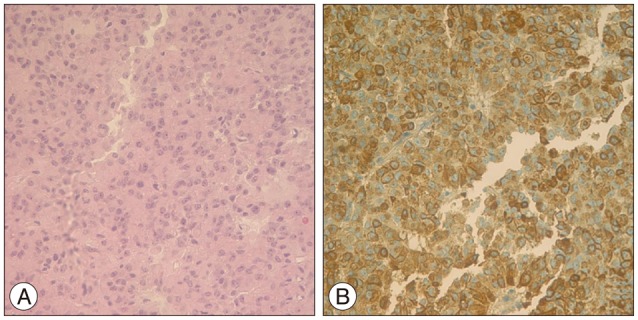
For the diagnosis of acromegaly by pituitary adenoma, he underwent endoscopic transsphenoidal tumor removal under navigational guidance (Fig. 3). In the operative field, we found abnormal pituitary mass, which was localized to left side and removed tumor. Postoperatively, hGH and IGH-1 were gradually decreased from 10.39 to 1.65 in hGH and from 838.3 to 704 in IGH-1. The GNAS gene (pseudohypoparathyroidism, type 1a), which is somatic mutations in GNAS, encoding the α-subunit of the heterotrimeric G protein complex (Gsα), occur in fibrous dyplasia and McCune-Albright syndrome b was positive in pituitary tissue and negative in serum specimen.
DISCUSSION
In this case, the patient had polyostotic (skull, left mandible, and left iliac bone in bone scan) and GH secreting pituitary adenoma, but did not display café-au-lait macules compatible with a diagnosis as MAS. In addition, genetic mutation about GNAS gene (pseudohypoparathyroidism, type 1a) in pituitary tissue was positive. In almost every prior case reported the base of the skull was involved, as seen in our patient.
MAS is a genetic abnormality that is an activating mutation in the GNAS1 gene that maps to chromosome 20q13 and encodes a ubiquitously expressed stimulatory (Gsα) subunit of the G protein11). Point mutations of Gsα result in constitutive activation of adenylyl cyclase and high cAMP levels with resulting increased mitogenic signaling and autonomous hyperfunction of several endocrine glands. The same activating Gsα mutation is in the bone affected by fibrous dysplasia10).
In our case, GNAS gene mutation in pituitary tissue was positive, but negative in serum specimen. However, this was not important for diagnosis because MAS manifests unusual genetics in that it is a postzygotic (non-germline) mutation resulting in both genetically-normal and abnormal cells (mosaicism) being present throughout the body1,11).
Optimal current treatment for GH secreting pituitary adenoma is surgical resection. Even subtotal resection of hypersecretory adenoma is the effective initial therapy to reduce excess hormone levels7). In MAS patients, however, even subtotal pituitary adenoma resection is often prohibited by severe skull base fibrous dysplasia. The transfrontal approach or combination of transsphenoidal and transfrontal approach has been required in some cases6).
There are some differences between our case and other prior reported cases. First, in our case, initial endocrine evaluation was normal finding and only fibrous dysplasia in the skull was evident. However, after 2 years of diagnosis of fibrous dysplasia, the patient had prominent acromegaly feature and showed increased hGH and IGH-1 levels in endocrine evaluation. We did not check the MR images initially because of no acromegaly feature and no abnormalities in hormone evaluation. In some patients with fibrous dysplasia, MR images will be helpful for evaluation of hormone secreting pituitary adenoma.
The first operation was done to decompress the left optic canal for symptom of left eye visual disturbance. The second operation was to remove the hGH secreting pituitary tumor. The second operation, transsphenoidal tumor removal, was complicated by distortion of anatomy by the bony hypertrophy and prior operation. In almost all prior MAS cases, there were remnant tumors after TSA operation. However, in this case the total pituitary tumor was removed and the patient has not experienced any neurologic change.
 XML Download
XML Download