

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic inflammatory demyelinating polyneuropathy (CIDP) is an acquired peripheral neuropathy that affects motor and sensory nerves and usually leads to perivascular endoneurial infiltration by monocytes, macrophages, and lymphocytes8,24).
Charcot-Marie-Tooth disease (CMT) is a genetic disorder that affects the peripheral motor and sensory nerve like as CIDP38,39). In CMT type 1 (CMT1) which is the most frequent type of CMT, patients are young at the onset of the disease, and it progresses very slowly. CIDP, 1-2 per 100000 adults28), usually affects the elderly and shows a rapid progression with a relapsing or progressive course21,31,36). Early-onset CIDP is rarer, 0.5 per 200000 children6,28,30), and progresses more slowly31,32).
The prognosis and response to treatment are different between CMT1 and CIDP40), and it is crucial to distinguish one from the other3,31). Although the hypertrophic cauda equina is uncommon in both CIDP and CMT14,11,17,23,35), it sometimes needs surgical treatment for cauda equina syndrome (CES)11,12,17,23,37).
Here, we report a rare case of early-onset CIDP with CES, which was clinically similar to CMT, and review the relevant literature.
CASE REPORT
A 34-year-old man presented to our department with low back pain, radiating pain, and weakness of bilateral lower extremities, which had progressed slowly during 3 months, despite medication and physiotherapy. He could not walk and needed a wheelchair to ambulate because of flaccid paraparesis. He also had intermittent urinary incontinence and constipation.
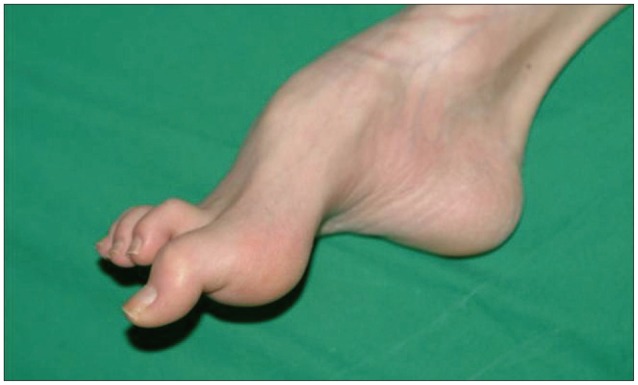
He showed muscle atrophy in the bilateral lower extremities and a thin body shape. He had foot deformities, hammertoes and pes cavus, since age 10, which made him to walk on his toes (Fig. 1). His family history did not include any incestuous marriages or patients with similar symptoms.
Both lower extremities showed grade 4 weakness. Sensations of pain, temperature, vibration, and proprioception were decreased below the L2 dermatome. The patient showed reduced knee and ankle jerk reflexes on both sides, but no pathologic reflexes.
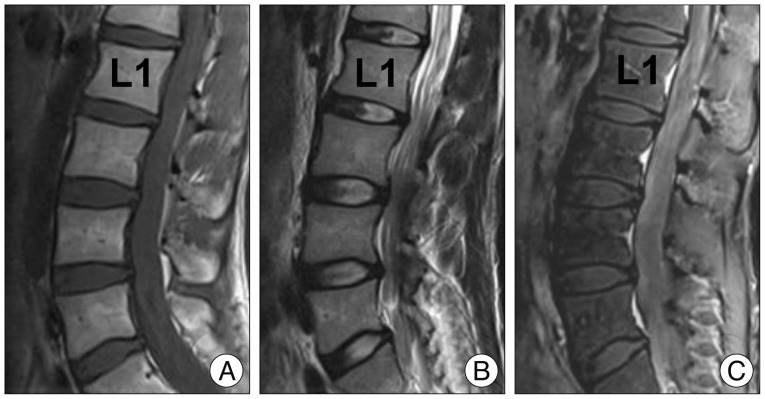
The spinal MRI revealed a diffuse thickening of the cauda equina, resembling an intradural tumor. This showed low signal intensity in both T1 and T2-weighted images, and slightly inhomogeneous high signal intensity on T1-weighted gadolinium-enhancement (Fig. 2). The brain MRI findings were normal. Hematologic tests for tumor markers (AFP, CEA, PSA, TFT), autoantibodies (ANA, ANCA, SS-A), and viral antibodies (VDRL, HTLV, HIV) were all negative.
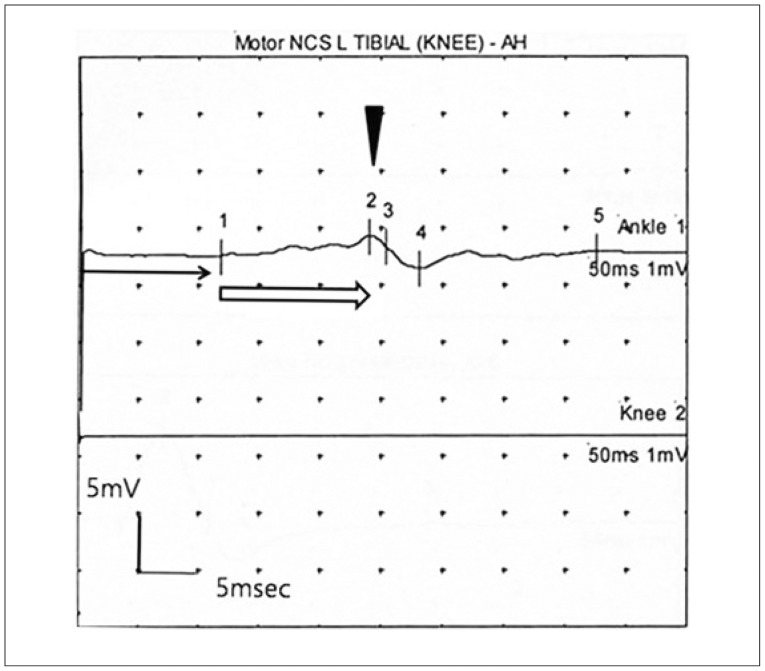
On electrophysiological tests, both upper and lower extremities showed conduction blocks, increased distal latency, temporal dispersion, severe reduction in nerve conduction velocity (NCV, 3 m/s), prolonged F-wave latency, and decreased signal amplitude on tibial nerve (Fig. 3). Electromyograms of the lower extremities showed positive sharp waves in the bilateral first dorsal interosseus, vastus lateralis, tibialis anterior, and gastrocnemius muscles. These data suggested a severe, demyelinating-type sensorimotor polyneuropathy.
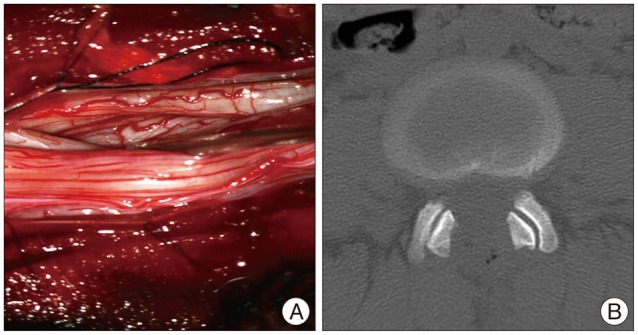
The patient underwent L2-5 decompressive laminectomy and sural nerve bisopy. Clear cerebrospinal fluid gushed out when the dura was opened, and unusually thick nerve roots were visible. CSF sampling was done. Exploration of the intradural space revealed hypertrophied nerve roots with size variation from normal to about treble size (Fig. 4). Then the dura was closed without patch duraplasty because the intradural space seemed to be enough for the swollen roots by virtue of the extra space provided by the decompressive laminectomy. In sural nerve biopsy, light and electron microscope examinations revealed myelinated axon surrounded by proliferated Schwann cells, 'onion bulbs' (Fig. 5). These pathological findings were compatible with chronic demyelinating peripheral neuropathy with remyelination, which can be seen in CMT1 and CIDP31,39,41).
The CSF examination showed normal findings. We performed a genetic examination at autosome 17p12-p11.2 (the PMP 22 gene) and the genes for MPZ, PRX, and EGR2, which frequently contain mutations in CMT patients38). However, we could not find any mutation.
After surgery, the patient showed immediate and marked improvement in pain, sensory deficits, urinary incontinence, and constipation. But, his motor power did not improve immediately after surgery. The patient received high dose intravenous IgG (IV-IgG) (0.4 g/kg/day) for 5 days and oral steroid (prednisolone, 1 mg/kg/day) for 1 week after the surgery. His motor power improved rapidly with the medications, and he could walk independently at 1 week after surgery.
Repeat physical examination and electrophysiological testing performed 1 week after the initial IV-IgG and steroid treatment course showed more quickly improvements in sensory and motor responses. High dose IV-IgG was initiated and followed by monthly for 3 months, but we used once for initial 5 days in our case because high cost36). After discharge, oral steroid therapy (prednisolone, 1 mg/kg/day) was maintained for few weeks and then tapered gently over months36). At one month after discharge, he showed normal motor power of both legs. His foot deformities were not changed. There was no relapse or aggravation of disease during 3 years of follow up.
DISCUSSION
Hypertrophy neuropathy that may induce hypertrophic cauda equina include CIDP11,17,23), Guillain-Barré syndrome7), malignant lymphoma20), paraneoplastic syndrome25), sarcoidosis13), CMT4,35), postirradiation neuropathy13), and various infectious diseases13). In the current case, we suspected the patient suffered from CIDP or CMT according to his symptoms and lab results. Both CIDP and CMT induce demyelinating polyneuropathy of the peripheral nerves, and thus, they may present similar symptoms, such as lower extremity motor weakness31). But, early differential diagnosis is necessary because of different prognosis and response to medication between CIDP and CMT15,16,42).
CIDP is an acquired paralytic disease with high morbidity. It was first introduced to the literature in 19759). The cause of CIDP is unknown, but recent studies support a pathogenesis suggesting an autoimmune disease, possibly an acquired demyelinating neuropathy due to an autoimmune attack on the myelin proteins18). It has various clinical manifestations, but its main symptoms are symmetrical sensory deficit and motor weakness27). Also, although rare, it may present pure motor or pure sensory symptoms, tremors, ataxia, and in some cases, foot deformities33). Hypertrophic cauda equina arising from CIDP is quite uncommon11,17,23). This disease may arise at any age, but it usually affects adults with a progressive course over just a few months27,31). The subsequent course can be relapsing and remitting or chronic progressive course with intermittent relapsing21,36).
CMT is a genetic disorder that affects the neuromuscular function. It can be classified into 4 types (CMT1, CMT2, CMTX, CMT4) according to inheritance pattern, molecular diagnostic test, electrophysiological examination, and neuropathology38,39). CMT1 is most common type. Clinical manifestations of CMT1 usually include muscle weakness, pes cavus and hammer toe by demyelination of peripheral nerves31). As in CIDP, hypertrophic cauda equina is quite uncommon in CMT14,35). The disease progresses slowly and usually occurs during the first decade of life3,31,39,40). Indeed, early-onset CIDP is very rare and similar to CMT1 because of its early onset, slower progression, and foot deformities10,31). So, CMT1 seemed to be more likely the diagnosis of our case because his symptoms included pes cavus, early symptom onset (age 10), and a slow progression (over 20 years). However, the findings of lower extremities were not specific for diagnosis, and may arise in other diseases (peroneal muscular atrophy, Friedreich's ataxia, myelodysplaisa, poliomyelitis and so on) can cause foot muscle atrophy2).
Electrophysiological testing is another method for differentiating CIDP from CMT1. Generally, the electrophysiological characteristics of CIDP are as follows : 1) Severely, slowed NCV, less than 10 m/sec; 2) Conduction blocks and/or temporal dispersion of compound muscle action potentials; 3) Prolonged distal latencies; and 4) prolonged F-wave latencies1,14). However, NCV in CMT1 shows mild to moderate slowing of nerve conduction, less than 38 m/sec, with no signs of prolonged distal latencies, conduction block, temporal dispersion, or prolonged F-wave latency39). In our case, the patient's test findings were consistent with CIDP, showing increased distal latency, conduction block, temporal dispersion, prolonged F-wave latency and severe NCV slowing (3 m/s).
In CIDP, a CSF study will show elevated protein level, to more than 45 mg/dL26). However, CSF proteins generally show normal levels in CMT119). In our case, the CSF showed normal protein level, as in CMT1. However, it is hard to differentiate CIDP from CMT1 by examining the CSF, since protein levels may be normal in early-onset CIDP31).
The pathological hallmarks of CMT1 are segmental demyelination and consequent remyelination, the so-called onion bulb22). Demyelination and edema due to inflammatory infiltrates are the key characteristics of CIDP34). Hence, inflammatory infiltrates may indicate CIDP, whereas prominent onion bulb findings likely indicate CMT141). However, onion bulbs without inflammatory infiltrates can be seen in CIDP patients41). Therefore, differential diagnosis of the two diseases by nerve biopsy alone would be insufficient1). In our case, we observed typical onion bulbs without inflammatory infiltrates.
CMT is an inherited disease with family history, whereas CIDP is an acquired disease. Most of CMT cases are reported to have genetic mutation38), and it is important for differentiation from CIDP3,31,32). But, we could not see gene mutations in our case for CMT, i.e., PMP 22, MPZ, PRX and EGR2 genes5,38). And we thought the diagnosis of our case more likely as CIDP based on no family history and genetic mutation21).
Corticosteroids, IV-IgG, plasma exchanges, and immunosuppressive drugs have been used in treating CIDP29). Most forms of CIDP show a good response to steroid or IV-IgG, whereas CMT1 does not32). The prognosis of CIDP is variably depends on the response to the treatment15,16). There have been limited treatment options for CMT1, exercise, orthosis, pain control and some other medications, which did not show significant benefit42). Good therapeutic response to steroids or IV-IgG in CIDP is a useful differentiating point from CMT13,31,32). In our case, favorable response for steroid and IV-IgG might support a diagnosis of CIDP.
Negative gene study, no family history, electrophysiologic test, and good response to steroid and IV-IgG indicated higher possibility of CIDP. In addition to the findings supporting diagnosis of CIDP, we could confirm the diagnosis of early-onset CIDP according to the atypical clinical findings for usual CIDP, early onset, slower progression, and foot deformities.
Motor and sensory deficits due to inflammation respond well to steroid therapy, but CES due to hypertrophic cauda equina shows poor responsiveness to medication37). Hypertrophic cauda equina is known to often result in CES12,17,23,37). In CIDP with CES, decompressive laminectomy is effective for motor and sensory dysfunctions11,12,17,23,37). According to previous reports, surgical decompression for hypertrophic nerve root in CMT1 is also effective4,35). Our case showed prompt recovery from CES after decompressive laminectomy.
CONCLUSION
Clinical presentations of early-onset CIDP are similar to those of CMT1. However, it is important to distinguish one from the other because the prognosis and responsiveness to medical treatment differ between them. CES due to hypertrophic cauda equina is a good indication for early decompressive laminectomy.

 XML Download
XML Download