

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Stroke is the first leading cause of death with 10.4 percent incidence among single diseases in Korea33). More serious problem is the survivors which have cerebral infarction with disability. These survivors incurred multibillion annual dollar cost in America5). In the incidence of stroke, non-hemorrhagic infarct (ischemic stroke) was presented over eighty percent. Moreover, infarct damage within 3 hours from focal ischemia is the most important factor which determines survivability and disability of ischemic stroke.
Ischemic stroke reveals two separate areas with ischemic core and penumbra. The ischemic core indicated irreversibly injured tissue even though reperfusion was conducted2,15). Thus, most studies focused on ischemic penumbra which has possibility of recover from apoptosis. Although, the ischemic penumbra could be recovered by various therapies, the infarct volume with ischemic core and penumbra may be decided in the early stage of ischemic stroke already.
In the mechanism of ischemic stroke, excitotoxicity was considered a main cause of neuronal death. The excitotoxic mechanism was processed with excessive glutamate release, calcium influx, uncontrolled membrane depolarization, decline of cellular ATP and oxidative stress simultaneously1). In early stage of ischemic stroke, cytoplasmic calcium overload is a major excitotoxic event which was regulated by mitochondria calcium homeostasis. And, mitochondrial dysfunction was induced by increase of free radicals, in turn, diminished the antioxidant activity25,27). Thus, oxidative stress could be considered one of those important causes in apoptotic or necrotic neuronal death with DNA fragmentation, cell membrane disruption and blood brain barrier broken.
Carnosine is an intrinsic antioxidant expressed in various organ tissues including brain3,7,14). In the studies of neuroprotective activity, carnosine showed several functions of antioxidant including membrane protecting properties, complexing of heavy metal ions. The antioxidant capacity was related to glutamate release, membrane degradation, superoxide dismutase activity and ischemic penumbra. Indeed, exogenous carnosine administration induced decrease of lipid peroxidation, membrane disruption and oxidative damages by crossing the blood brain barrier9,16,24,29). Most studies of carnosine were conducted to investigate revitalization ability in ischemic penumbra after reperfusion. However, the early ischemic injury determined the volume of ischemic core and penumbra essentially11,19).
Therefore, in this study, we investigated the neuroprotective effect of carnosine in early stage of focal ischemia. The effect of carnosine was evaluated with antioxidant capacity and infarct volume.
MATERIALS AND METHODS
Materials
The neuroblastoma cell line (SK-N-MC) was purchased from ATCC (Manassas, VA, USA). The cell culture media of glucose free DMEM and dialyzed FBS were obtained from Invitrogen (Rockville, MD, USA). The reagents including carnosine, DCF-DA (2',7'-dichlorodihydrofluorescein diacetate), TTC (2,3,5-triphenyltetrazolium chloride) and antimycin A were obtained from SIGMA-ALDRICH (St. Louis, MO, USA). Antibodies of superoxide dismutase (SOD)-1, growth associated protein (GAP) 43 and GPADH were used from Cell Signalling Technology Inc. (Danvers, MA, USA). All commercial ELISA kits including SOD, CAT (catalase) and GPx (glutathione peroxidase) were purchased from Biovision (Milpitas, CA, USA), Cell Biolabs Inc. (San Diego, CA, USA) and Dojindo (Kumamoto, Japan).
Chemical hypoxia, cell survivability and ROS measurement
The neuroblastoma cell was cultured 10% fetal bovine serum at 37℃ in 5% CO2 incubator. When the cells reached confluence, culture media was changed with chemical hypoxia induction media (10 uM of antimycin A and 5% of dialyzed FBS in glucose free DMEM). Cell survivability was measured after 2 hours from chemical hypoxia induction with Automatic Cell Viability Analyzer (Beckman Coulter, Brea, CA, USA). And, reactive oxygen species (ROS) was expressed by 10 uM of DCF-DA for 5 minutes. The expression was observed with IX-70 fluorescence microscope (Olympus, Tokyo, Japan).
Animals and drug treatment
All experiment using animals were conducted in accordance with Guide for the Care and Use of Laboratory Animal from the National Institutes of Health (NIH). Carnosine was dissolved with 100, 250, 500 mg/mL in sterile saline. Rats with vehicle and carnosine treated groups were administrated by intraperitoneal injection before 30 minutes of surgery (n=5 per group).
Focal ischemia stroke model
The method of middle cerebral artery occlusion was adapted from Longa et al.21) and Kulik et al.18). Rats were anesthetized with isoflurane (3% for induction and 2% for surgical procedure) in 70% of N2O and 30% of O2. The rectal temperature was preserved between 36.5℃ to 37.5℃ with heating lamp and blanket. After a left side neck incision, the left carotid artery was separated without vagus nerve injury. A 4-0 nylon monofilament coated with silicone rubber was injected into the left internal carotid artery. The middle cerebral artery occlusion was maintained with ligation of nylon tread on the filament injection site. All of surgical procedure was completed within 15 minutes. The rats were euthanized 2 hours after ischemia with 150 mg/kg pentobarbital sodium by intraperitoneal injection. The brain and blood was immediately collected to measurement of infarct volume and analyses of neuronal factors.
Infarct volume measurement
The brain was cut into six coronal sections of 2 mm thickness using rodent brain matrix (JEUNG DO BIO & PLANT CO., LTD, Seoul, Korea). The brain pieces were stained with 2% TTC in phosphate buffered saline (PBS) for 1 hour at 37℃. After wash twice with PBS, sections were fixed with 10% phosphate-buffered formalin for 24 hours. The digital image of brain sections were generated using EPSON Stylus TX130 (EPSON Korea Co., Ltd, Seoul, Korea), and the infarct volume was measured using NIH Images J analysis software (version 1.44). The corrected infarct volume was calculated as described by Kim et al.17) : corrected infarct area (mm2)=total area of the contralateral hemisphere--intact area of the ipsilateral hemisphere. A total volume (mm3) of infarct was calculated by sum of each area of sections with thickness.
Western blot
The brain tissue including infarct core and penumbra was isolated simultaneously with euthanasia. After washing with PBS, protein extraction solution (Pro-Prep, Intron Biotechnology, Seongnam, Korea) was added 600 uL of lysis solution per 10 mg of tissue. According to homogenization with Pro2000 (Pro Scientific, Oxford, CT, USA), the homogenate was incubated in -20℃ for 20 minutes. With the remnant was removed by centrifugation, the concentration of total protein in supernatant was measured with Bio-Rad protein assay kit (Hercules, CA, USA). After the separation of the protein in 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), the protein was transferred to nitrocellulose membranes (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membrane was blocked with 5% of skim milk for 1 hour. During the detection process of the membrane, antibodies of SOD-1, GAP43 and GAPDH were used. The quantification of protein expression was calculated by Chemi-Smart 5000 (Vilber Lourmat, Marne La Vallee, France).
Enzyme-linked immunosorbent assay
According to the protocol of each ELISA kit, protein of brain was dissolved in DMSO or distilled water. In the analysis of antioxidant activity including GPx and CAT was conducted with manuals of Glutathione Peroxidase Activity Assay Kit and Catalase Activity Assay Kit (BioVision, Mountain View, CA, USA). And, SOD was analyzed by SOD activity assay kit manual (Dojindo, Kumamoto, Japan). The quantifications of activity were calculated by semi-quantitative methods from each standard.
Statistical analysis
The statistical analysis was conducted using Statistical Package for the Social Science (SPSS). The comparison of infarct volume, antioxidant activity and apoptosis related factor expression between control and carnosine treatment group was analyzed by chi-square test. A statistically significant difference was indicated by a p<0.05.
RESULTS
Neuroprotective effect of caronsine in chemical hypoxia in-vitro model
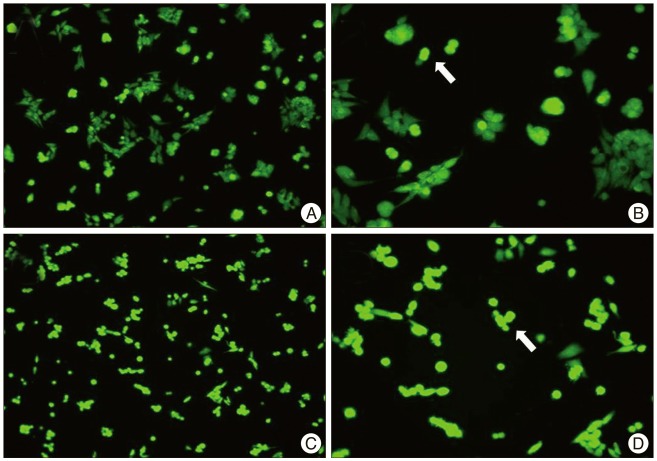
Chemical hypoxia was induced by 10 uM of antimycin A with glucose free media. With the increase of carnosine concentration, neuroblastoma showed higher cell viability in chemical hypoxia (Fig. 1). And, the significant difference was detected in over 10 mM of carnosine treatment group. In the comparison of ROS production with DCF-DA, carnosine induced decrease of ROS expression (Fig. 2).
Neuroprotection of carnosine in early stage of stroke model
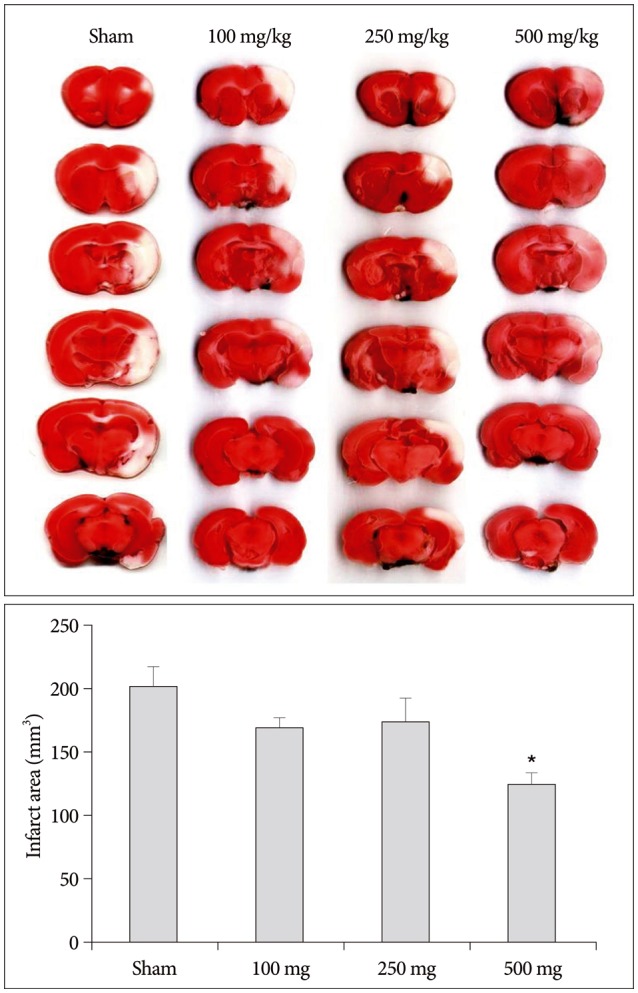
The neuroprotective effect of carnosine was investigated with coronal sections which were obtained by brain matrix at distances of 4, 6, 8, 10, 12, and 14 mm from the rostal extremity of rat frontal cortex after 2 hours of MCAo (Fig. 3). In early stage of MCAo model, average infarct volume was 201 mm3 areas. Intraperitoneal injection of 100, 250, and 500 mg/kg carnosine at 30 minutes before MCAo reduced the infarct size by 15.4% (170.30±18.03), 13.49% (174.17±44.83), and 38.16% (124.50±22.42; p<0.05) respectively. Indeed, the 500 mg/kg carnosine treated group showed expansion of distinguishable penumbra.
Effects of carnosine on serum antioxidant capacity
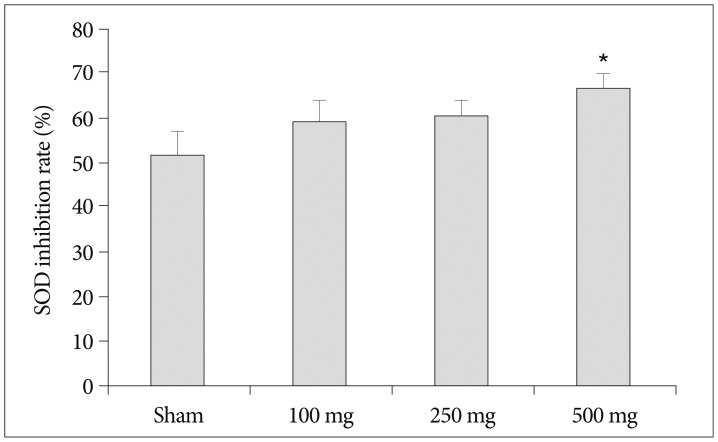
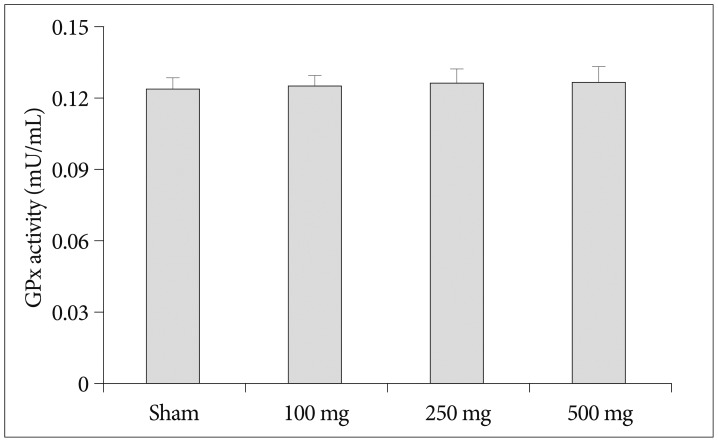
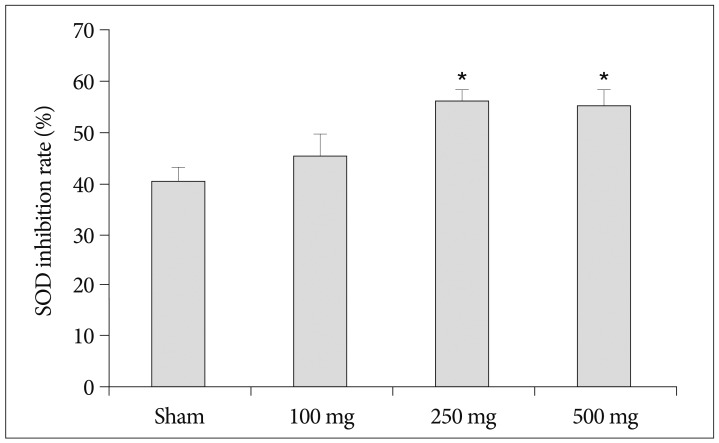
To determine whether the neuroprotective effects of carnosine are related antioxidant capacity, we investigated the effects of carnosine on serum SOD, catalase and glutathione peroxidase activity. As shown in Fig. 4, pretreatment of carnosine increase serum SOD inhibition rate following increase of carnosine concentration. Although, the SOD activity gradually enhanced by increase of carnosine concentration, the significant difference was detected only in 500 mg/kg carnosine treated group similar to the reduction of infarct volume. In the analysis of catalase, on the contrary, increase of carnosine concentration induced reduction of catalase activity (Fig. 5). And, carnosine 100 mg treatment group showed the highest activity significantly. However, the expression of GPx did not changed by carnosine treatment (Fig. 6).
Effects of carnosine on brain tissue
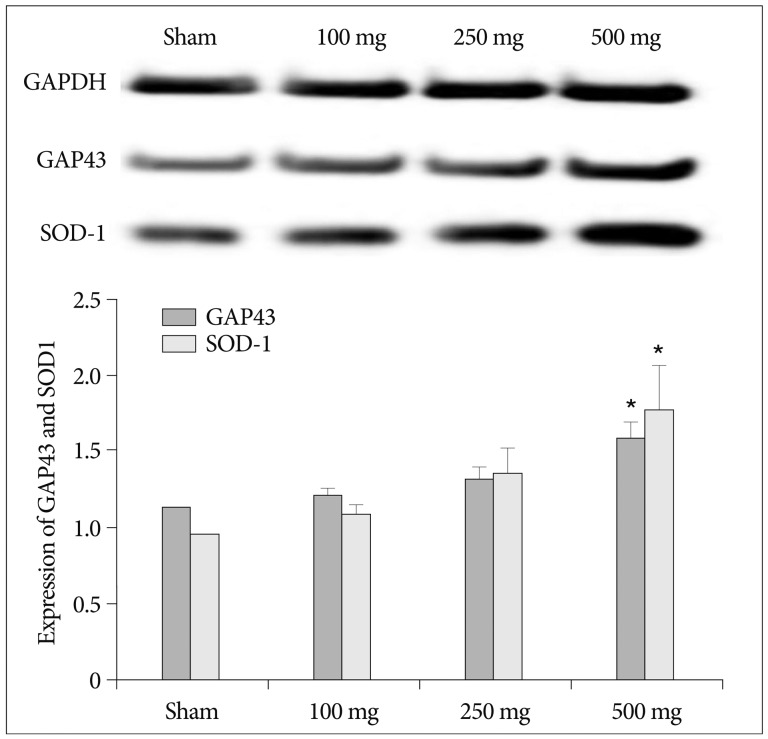
Carnosine also produced an increase in tissue SOD activity. Pre-treatment of 250 mg and 500 mg carnosine enhanced SOD expression in brain significantly (p<0.05) (Fig. 7). In the analysis of GAP43 and SOD-1, carnosine induced increase of cell survivability and SOD antioxidant expression (Fig. 8). Although the expression of GAP43 and SOD-1 increased in dose dependant manner, the significant difference was detected only in 500 mg of carnosine pretreatment group (p<0.05).
DISCUSSION
Neurons in the ischemic core of infarction were induced cell death within minutes15). And the ischemic penumbra surrounded the ischemic core with electrically silent2). In human, it is difficult to verify the division mechanism of ischemic core and penumbra in early stage of stroke. In this point of view, focal ischemia model could be considered as an appropriate model of permanent occlusion of the middle cerebral artery with a better representation of naturally occurring stroke29). In this study, the result of infarct volume was about 200 mm3 in 2 hours pMCAO models. Although there was no similar experiment to comparison of previous studies, the ischemic core and penumbra might be determined within 2 hours of MCAO20). In early stage of infarct model, carnosine pretreatment showed not only a significant reduction of ischemic core, but also increase of penumbra. And, the analysis of GAP43 which was neuronal marker supported the neuroprotective capacity of carnosine. These results mean two advantages with reduction of a physical disorder after surgery and extension of time limit for surgery. Since the penumbra has a potential to recovery with collateral blood reperfusion13,23).
In early stage of stroke, the major injury is caused by decrease of oxygen and glucose supply. Mitochondria of neuronal cell in brain consumed 90 percent of oxygen to generate ATP. Induction of apoptosis by chemical hypoxia with 10 uM of antimycin A within 2 hours indicated that mitochondria dysfunction may important factor in injury of stroke. Indeed, neuronal cell death and ROS production was decreased by carnosine treatment significantly. Since, carnosine related anti-ischemic activity, antioxidant and membrane-protecting properties32). With the ATP depletion by chemical hypoxia, mitochondria dysfunction is induced by oxidative and nitrosative stress with free radicals. The results of mitochondria dysfunction induced decrease of the antioxidant capacity and trigger of apoptotic cascasde4,25,26,27). Thus, carnosine may induce the decrease of oxidative stress which was associated with blood brain barrier disruption, excitotoxicity and ischemic injury in early stage of stroke.
In the antioxidant cascade, there are two kinds of antioxidant which were enzymatic and non-enzymatic components. The enzymatic antioxidants were a primary intrinsic antioxidant with superoxide dismutase (SOD)12), catalase (CAT)22) and glutathione (GPx)10). And, the non-enzymatic antioxidants were small molecules including ascorbic acid (vitamin C), alpha tocopherol (vitamin E) and tripeptide glutathione (GSH) which have scavenging activity of free radicals28). In the analysis of antioxidant expression, there are different pattern following carnosine treatment dose. Although SOD expression in blood showed similar pattern to protection of infarct, CAT expression showed the highest increase in low dose of carnosine treatment. In this result, there are two presumptions with time dependent manner and action of carnosine low dose. Since, CAT expression in low dose of carnosine showed significant increase against control. Indeed, the SOD showed consistent pattern in brain and blood with increase of SOD-1 which was copper/zinc SOD. These results indicate that carnosine protect brain injury with enhance of antioxidant capacity in early stage of stroke.
In an aspect of clinical usefulness, carnosine have several advantages for clinical application. Carnosine is a naturally-occuring non essential amino acid in human body. And, the supplemented carnosine is absorbed into plasma intact8). Indeed, previous studies suggested that the applicable dose of carnosine supplementation was 1.6--6.4 g/day in human including 800 mg/day in children6,31). In this study, effective dose of 500 mg/kg carnosine in rat can be converted 81 mg in human with human equivalent dose (HED)30). It is ten-fold lower concentration than that in clinical test of children with 800 mg. Thus, carnosine could be considered as preventive medicine candidate with antioxidant capacity, supplementation efficacy and predictive low adverse effect.
CONCLUSION
The risk of stroke incidence consistently increased with aging of population. Thus, many studies was conducted to protect and recovery of brain injury. In this study, we demonstrated the neuroprotective effect of carnoine using early stage of stroke model. And, the action of carnosine was related neuronal cell survivability and antioxidant capacity in chemical hypoxia and ischemic stroke rodent model. Although, there are many risk factors in ischemic stroke including NMDA excitotoxicity, calcium influx and apoptosis signaling, carnosine induced significant decrease of brain injury in early stage of stroke. Thus, the protection of oxidative stress by mitrochondria dysfunction should be considered one of major risk factor in early stage of stroke.


 XML Download
XML Download