

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Malignant peripheral nerve sheath tumors (MPNST), which are comprised of 5 to 10% of all soft-tissue sarcoma, often occur in the near proximal extremity at a nerve trunk such as sciatic nerve, brachial plexus, or sacral plexus. They have a distinct association with the neurofibromatosis type 1 and occur in 3-15% of NF1 patients and a high incidence of tumor is related to the mutation of the NF1 gene9). On the other hand, a primary malignant intracerebral nerve sheath tumor (MINST) is quite rare, with only 8 cases1,2,4,10,12,13,15,16) documented in English-language literature. We report on a case of MINST and the relevant literature is reviewed.
CASE REPORT
Clinical course
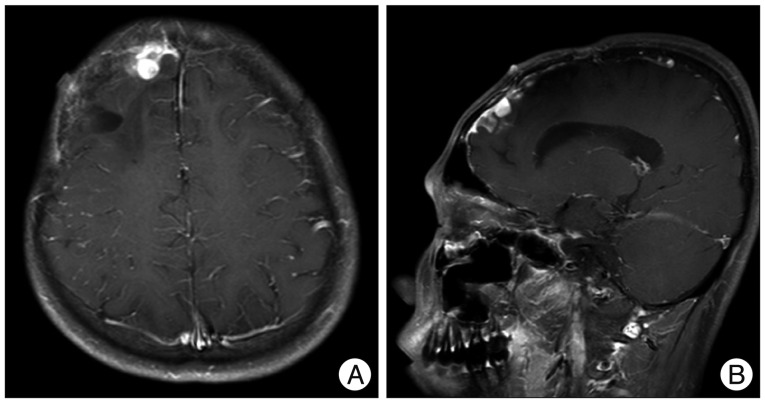
A 13-year-old boy was hospitalized due to the headache on the right side of his head, which began 2 weeks ago and the vomiting, which began a week ago. The patient was alert and showed no neurologic abnormalities. There was no evidence of neurofibromatosis such as neurofibroma, café au lait spot, Lisch nodule, optic glioma, distinctive bony lesion (dysplasia of the sphenoid bone or long bone cortex) and freckle in the groin or axilla with no family history of neurofibromatosis. Brain magnetic resonance imaging (MRI) revealed about 6.7 cm diameter, a well defined highly enhancing mass in the right frontal lobe (Fig. 1). The preoperative differential diagnoses were glioblastoma multiforme, anaplastic astrocytoma, and malignant meningioma. A surgical resection was carried out through right frontal craniotomy.
Diagnosis
Macroscopically, it was a well-circumscribed yellowish nodular round mass having gelatinous surface without dural attachment (Fig. 2). It was intra-parenchymal tumor and had a distinct cleavage plane in the superficial brain parenchyma, but not in the deeper portion. Microscopically, the tumor was highly cellular and mainly consisted of spindle cells in interlacing and interwoven fascicles. Perivascular whirling and condensation of tumor cells were observed. The tumor was well-demarcated from the adjacent brain tissues. Each tumor cell had a fusiform nucleus and aligned in a convoluted form with a moderate degree of mitotic activity and a high nuclear-to-cytoplasmic ratio. Myxoid parts were scattered in the tumor (Fig. 3).
Immunohistochemistry showed that the tumor cell was positive for S100 (Fig. 3), but negative for glial fibrillary acidic protein (GFAP), CD34, synaptophysin and desmin. These histological and immunohistochemical findings supported the diagnosis of a primary MINST.
Postoperative treatment
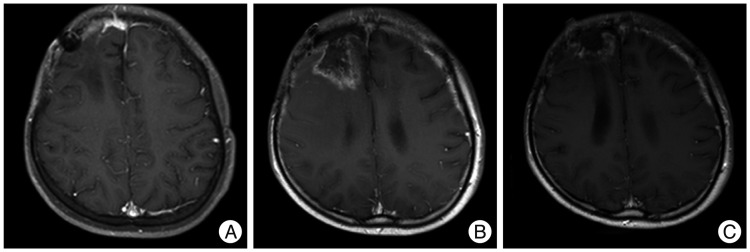
Thirty times of fractionated whole-brain radiation were performed with 5760 cGy over the period of 6 weeks after surgery. Brain MRI follow-up examination was performed every six months, but there was no evidence of tumor recurrence. Brain MRI at 50 months after his first surgery revealed a 1.0×1.0 cm of well-defined enhancing mass in the right frontal convexity and leptomeningeal enhancement in the right frontal area (Fig. 4). Whole spinal MRI showed no evidence of metastasis. No malignant cell was observed in his cerebrospinal fluid study. Second gross total resection (GTR) was performed. The histopathological examination showed a primary MINST, which was consistent with the histopathological results from the first surgery. The brain MRI at four months after second surgery revealed a tumor of 1.0×0.5 cm in size in the right anterior frontal convexity and pachymeningeal enhancement in the right frontal area (Fig. 5A). As the patient's guardian wanted to treat the patient in another hospital, so the patient was transferred to another hospital. Third GTR was carried out. The histopathological examination also showed a primary MINST. As the tumor was repeatedly relapsed despite the GTR which was performed twice before, radiation therapy and chemotherapy were performed according to the Korean Society for Pediatric Neuro-Oncology (KSPNO)-S081 Protocol, which can be used for a high-grade brain tumor13). Thirty times of fractionated radiation were carried out with 6000 cGy after his third surgery and 4 times of combination chemotherapy with vincristine (1.5 mg/m2 IV push, Day 0, 7), etoposide (75 mg/m2 in normal saline dose ×2.5 times IV over 2 hours, Day 0-4), carboplatin (300 mg/m2 in normal saline 125 mL/m2 IV over 1 hour, Day 0, 1), and ifosfamide (1500 mg/m2 in dextrose water 200 mL/m2 with Mesna (300 mg/m2 IV over 1 hour, Day 0-4) were applied. The irregular enhancing lesion (3.0×4.0 cm) was found in a brain MRI at six months after the third surgery (Fig. 5B). As the size of this lesion was found to be smaller than the previous one in the brain MRI at thirteen months after the third surgery, this lesion is considered to be the result of radiation injury rather than tumor recurrence (Fig. 5C). He is still alive at 77 months after his first surgery and complaining of mild headache and dizziness, but there are no other neurological abnormalities.
DISCUSSION
The primary MINST is termed as a result of their histological and immunohistochemical similarities to MPNST but is distinguished by their intracerebral location1). MINST is a preferable term to indicate malignant intracerebral schwannoma or neurofibrosarcoma, since these tumors have the appearance of any nerve sheath cells, including Schwann cells, perineural fibroblasts, or fibroblasts8,10). Patients with MINST have little probability of concomitant findings of NF116).
Neither our case nor the other previously-published case reports could make an accurate diagnosis before pathologic findings. In imaging studies such as brain computed tomography and MRI, it is difficult to discriminate between MINST and high grade glioma, because those tumors were intra-axial, homogeneously enhancing and had surrounding edema11).
It is difficult to make an accurate diagnosis of MINST by looking ordinary hematoxylin and eosin-stained sections alone. The differential diagnosis of these tumors includes malignant melanomas with Schwann-type differentiation, mixed gliomas with mesenchymal differentiation, intracerebral rhabomyosarcomas, desmoplastic infantile ganglioma, intracerebral meningioma, and solitary fibrous tumor2,16). Isolated immunospecific marker for nerve sheath tumors was nonexistent. However, immunohistochemical analysis has special value both in verifying nerve sheath derivation and malignancy16). It is essential for distinguishing MPNST from the other malignant spindle cell tumors12). Positive S-100 protein may help in differentiating the nerve sheath tumors from the other soft tissue tumors6). The absence of GFAP expression in tumor cells ruled out desmoplastic astrocytoma, gliofibroma, and gliosarcoma, whereas the absence of synaptophysin and neurofilament protein excluded desmoplastic ganglioglioma12).
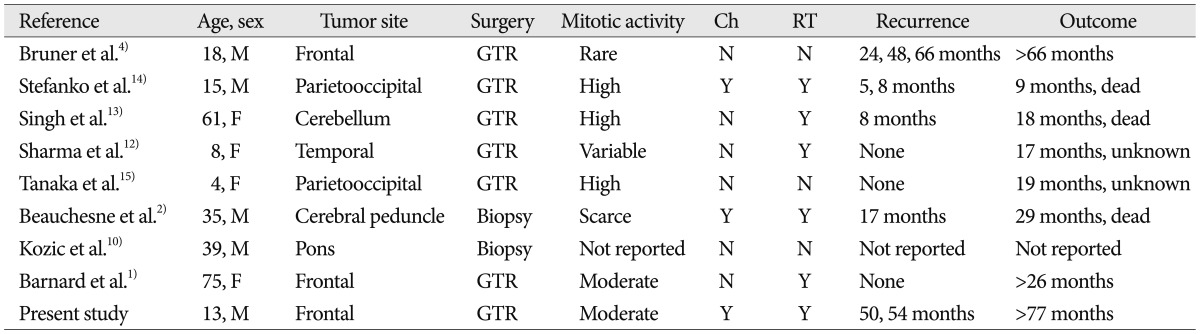
In the reported cases, the prognosis of MINST is poor2). According to the data in Table 1, survival time was longer when the tumor recurrence was slower. As the tumor recurrence became faster, the prognosis got worse. The recurrence free survival after the initial resection appears an important predictor of the overall survival12). In cases of Bruner et al.4), Barnard et al.1), and this case which reported long-term survival, the mitotic activity was rare or moderate rather than high. It seems that if the mitotic activity is low, the recurrence rate is also low and thus the survival time will increase, although we could not take into account of various factors due to insufficient data caused by few number of reports have been produced to date.
The extent of resection seems to be important in the prognosis of MPNST. A longer survival time was reported when total resection rather than subtotal resection was carried out3,7,16). Also, in MINST, GTR has always been proposed in the literature1,2,6,15).
It was considered that radiotherapy contributed to playing a limited role in the management of tumor in the early literature14,15). However, in the recent literature, radiotherapy showed possible effects on the local control and recurrence2,5), but little on the overall survival in patients with MINST1,16).
In case of Beauchesne et al.2), systemic chemotherapy with doxorubicin (3 courses every month) was prescribed, with mild benefit for the patient. However, because of the rarity of this tumor type, there are still few reports published and little available information about the chemotherapy8). In this case, the KSPNO-SO81 protocol was applied, which is one of protocols to treat a pediatric high grade brain tumor such as medulloblastoma and primitive neuroectodermal tumor. Although appropriate regimen of radiation therapy and chemotherapy are not determined yet2,5,15), it is considered to be useful for the long-term survival of the patient to carry out a surgery, adjuvant radiation, and chemotherapy, as shown in this case.
CONCLUSION
We report a long-term survival case of a primary MINST. Different from the previous cases, this is the first case that had no evidence of tumor recurrence for 50 months after first surgery, and showed long-term survival for more than 77 months without showing no evidence of tumor recurrence. Although a definitive therapeutic regimen for MINST has not yet been established due to the limited number of reported cases, a GTR followed by adjuvant radiotherapy and chemotherapy will be recommended to patients of MINST.



 XML Download
XML Download