

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pleomorphic xanthoastrocytoma (PXA), first described by Kepes et al.14), is a rare neoplasm that accounts for 1% of all astrocytic tumors. PXAs are supratentorial and involve the temporal lobe, so the most common presentation is seizure3,6,12-14). The histologic features of the tumor are marked cellular pleomorphism, variable lipidization and abundant reticulin. Features such as necrosis, nuclear atypia and mitotic figures are suggestive of a high grade tumor7,11,14,17,18,21). Despite this, PXAs possess a relatively favorable prognosis with a 10-year survival of 70%, and are classified as grade II in the World Health Organization (WHO) classification6,20). However, some PXAs recur and become malignant. In these cases, factors such as mitotic activity and surgical extent are known to be related to prognosis6). However, the clinical course and prognostic factors are still unknown. Here we describe the clinicopathologic and radiologic features, treatment outcomes and prognostic factors of 22 patients with PXA in a single institution.
MATERIALS AND METHODS
Between January 2000 and March 2012, 22 consecutive patients were pathologically diagnosed with PXA in our institute. A comprehensive analysis was conducted on these patients with regard to the clinical, radiologic and pathologic features at diagnosis as well as treatments and outcomes.
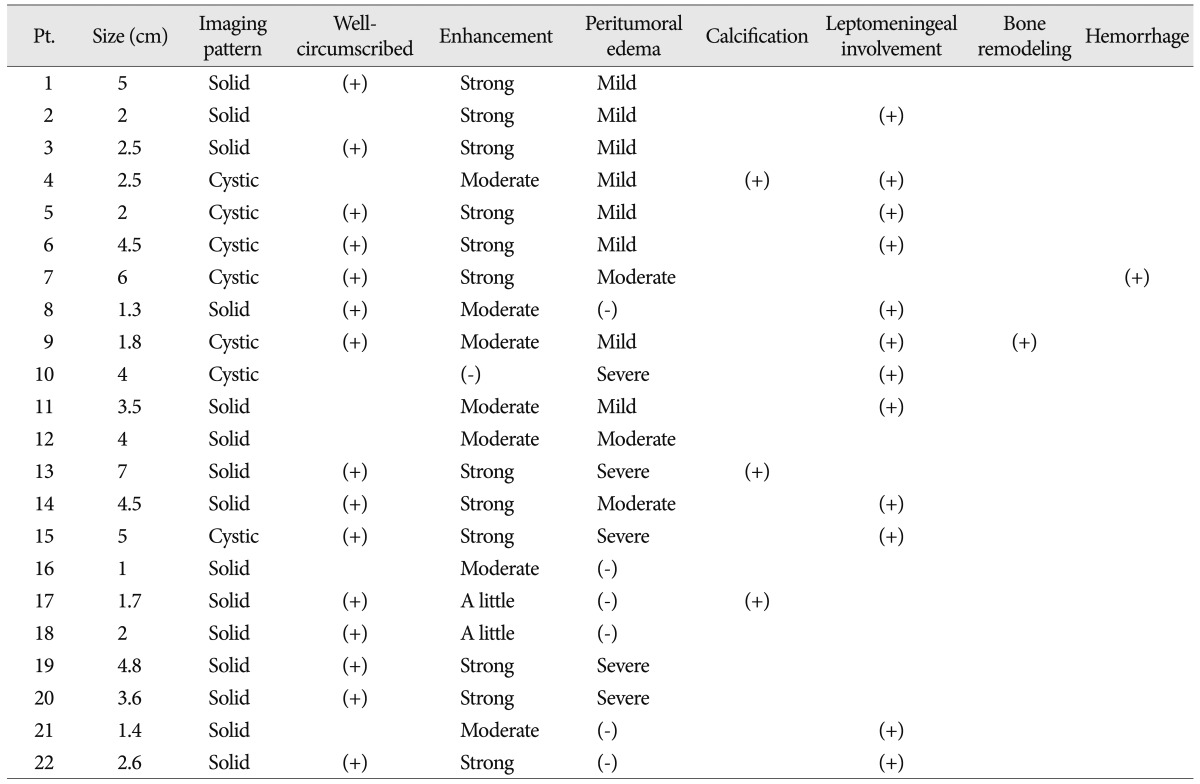
All patients underwent diagnostic and serial follow-up computed tomography (CT) or magnetic resonance (MR) scans on a regular basis. The images were evaluated with respect to the following factors : location, degree of enhancement, size, demarcation, presence of cystic degeneration, peritumoral edema, hemorrhage, calcification, leptomeningeal involvement and bone remodeling. Tumor size was determined as the longest dimension measured on gadolinium enhanced T1-weighted images. Large tumor was defined as its diameter greater than 3 cm. Peritumoral edema was defined as a maximum extent of increased T2 signal intensity on the tumor margin. Peritumoral edema was classified20) as mild, <1 cm; moderate, >1 cm; severe, >2 cm. An atypical tumor site was defined as any location other than the supratentorial cortex area.
A diagnosis of PXA was established based on the pathologic features described in the WHO classification of tumors of the central nervous system. The presence of necrosis, mitotic figures and Ki-67 were carefully evaluated by two pathologists.
The objective of surgery in all patients was to achieve gross total resection (GTR). The extent of resection was defined as follows : GTR, no surgical or imaging evidence of residual disease; near total resection, <10% residual tumor found in the surgical report and post-operative imaging; and subtotal resection (STR), <50% residual tumor found in the surgical report and post-operative imaging.
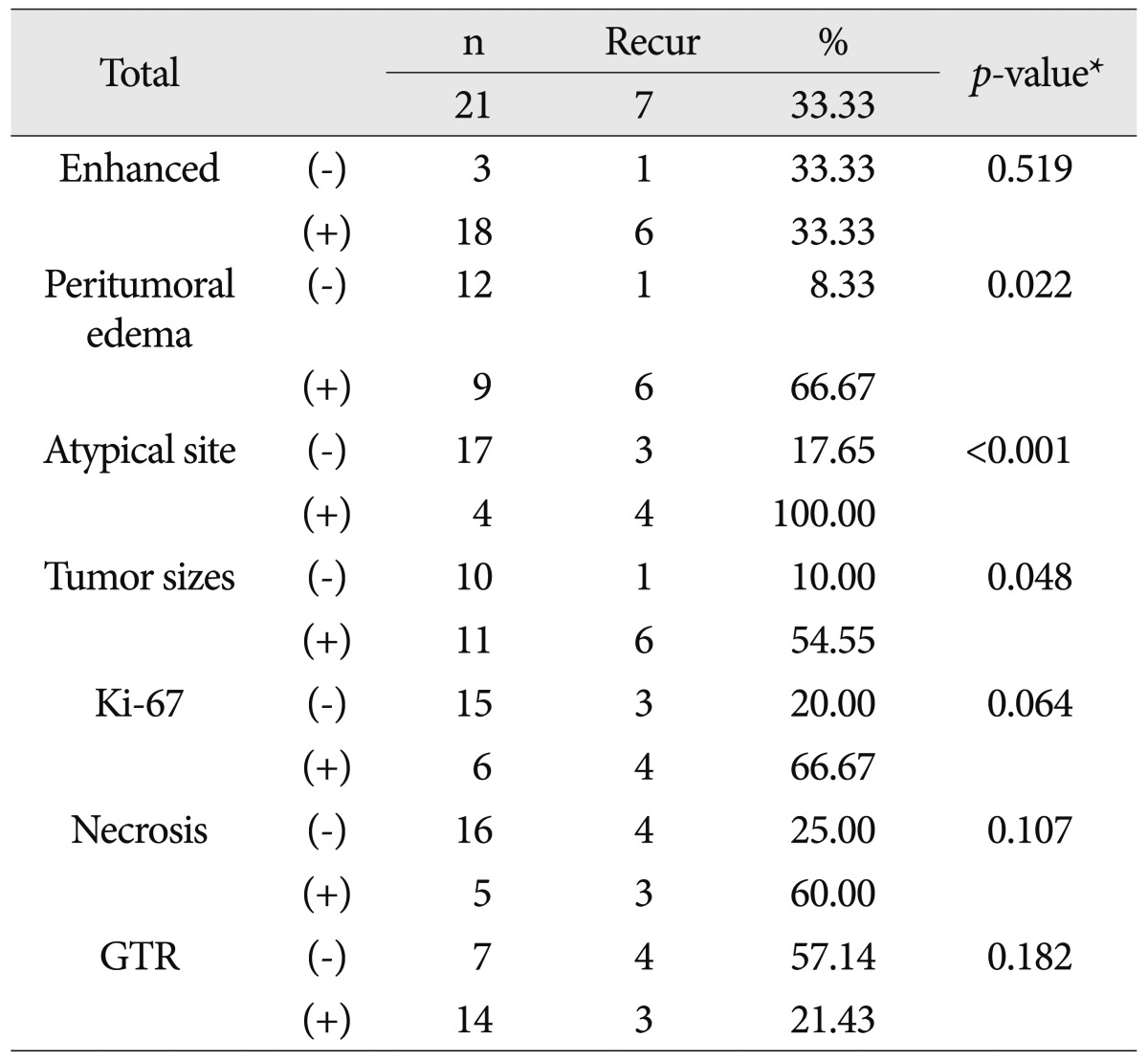
We evaluated the relationship between disease progression and enhancement, peritumoral edema, tumor size, atypical site, Ki-67 index, necrosis and GTR by univariate analysis. One patient who died from postoperative complications (brain edema) was excluded from the statistical analysis of overall survival and prognostic factors. Survival and local control were the primary analytic endpoints. Local control was defined as the absence of any tumor regrowth or progression on neuroimaging studies. Survival and local control rates were calculated with reference to the date of surgery (all patients underwent an operation as part of their initial treatment) using the Kaplan-Meier product limit method. Survival curves were constructed with Kaplan-Meier estimates and compared with the use of log-rank test. For all statistical tests, p<0.05 were considered significant.
RESULTS
Clinical characteristics of the 22 patients
Table 1 summarizes the clinical characteristics of the 22 patients included in the study. Male : female ratio was 9 : 13 and mean age was 23.8 years. Symptoms at presentation were seizures (n=14), headache (n=4), neurologic deficits (n=3) and dizziness (n=1). Apart from one patient who had a cerebellar tumor, all other patients presented with tumors in the supratentorial hemispheres. The primary tumors at typical sites consisted of temporal lobe (n=10), frontal lobe (n=4) and parietal lobe (n=3). The primary tumors in atypical sites were in the corpus callosum (2 patients) and lateral ventricle, thalamus, and cerebellum (1 patient each).
Radiological findings
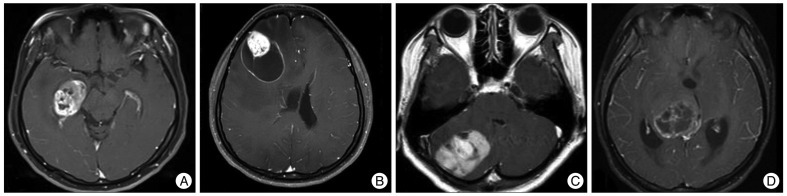
Neuroradiological findings are summarized in Table 2. Imaging revealed cystic dominant tumors in 7 patients and solid dominant tumors in 15. Most of the tumors were well-circumscribed and well-enhanced. In all, there were 12 were strongly enhanced, 7 moderately, 2 little enhanced and 1 not enhanced (Fig. 1). Fifteen tumors had clean margins in the brain parenchyma, but the remaining 7 did not. There were 11 large tumors and 10 small tumors and its size varied from 1 cm to 7 cm. Levels of peritumoral edema differed, with 6 displaying no edema, 8 mild edema, 3 moderate edema and 5 severe edema. Calcification was found in 3 patients, leptomeningeal involvement in 12 patients, bone remodeling in 1 patient and intratumoral hemorrhage in 1 patient.
Histopathological findings
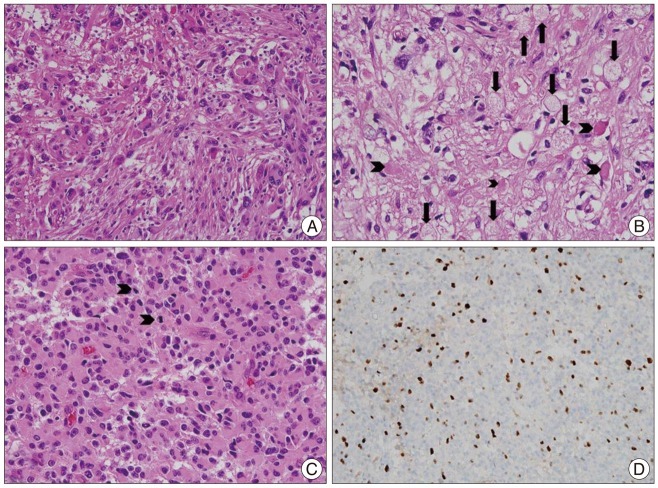
The tumors were well demarcated from the adjacent brain parenchyma. The tumor cells were pleomorphic, comprising spindle cells and giant cells with one large atypical nucleus and occasionally multiple nuclei (Fig. 2A). The nuclei of the giant cells had variable morphology, size and chromatin patterns with occasional intranuclear inclusions. The cytoplasm was amphophilic or eosinophilic. Some cells had small vesicles in their cytoplasm, creating a lipidized or xanthomatous appearance. Eosinophilic granular bodies were readily identified (Fig. 2B). Mitotic counts ranged from 1 to 8/10 HPFs (Fig. 2C). Ten tumors contained no mitotic figures and 3 tumors contained mitoses above 5. Necrosis was present in 5 cases (Table 3). The Ki-67 labeling index was >5% in six tumors (Fig. 2D). The tumor cells were positive for GFAP in all cases. Three (14%) of the 22 patients were initially diagnosed as PXA with anaplastic features, and one patient's tumor underwent malignant transformation.
Treatments and outcomes
The objective of the surgery in all cases was to achieve GTR. Initially, GTR was achieved in 14 patients, STR in six patients and a biopsy in one case. Reasons for the incomplete tumor resection were difficult location in five patients and poor demarcation in two patients. All seven patients who underwent STR were irradiated conventionally or by gamma knife radiosurgery (GKRS), and four of them underwent a second operation. One patient (No. 11) whose disease progressed after radiotherapy received adjuvant temozolomide chemotherapy.
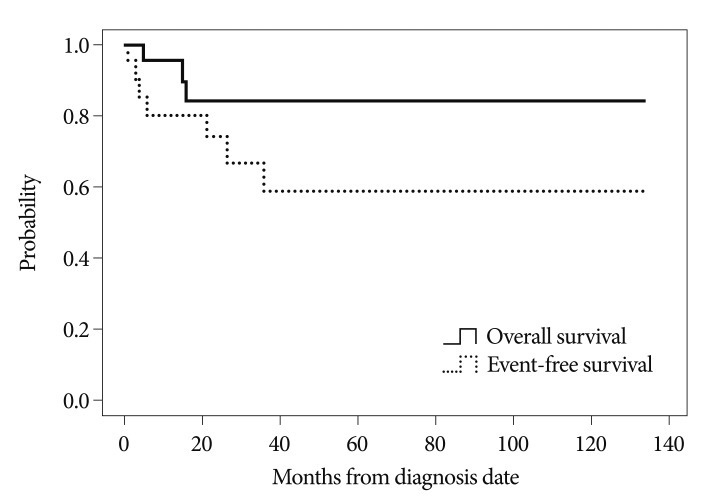
Follow-up data were available for 21 patients and follow-up ranged from 3 to 134 months (median : 31.0 months). Overall survival at 3 years was 84% with event-free survival of 59% (Fig. 3). Three patients (14%) died following progression of the disease; one at 3 months and two at 15 months. Of the 21 patients, 7 (33%) suffered disease progression. Metastasis occurred in two patients : in one (patient 2) the metastasis was in the intramedullary spinal cord with no evidence of CSF seeding (the spinal tumor was resected and the patient recovered), and in the other there was CSF seeding (patient 19, who died of the disease).
Three of the 14 patients who received GTR had recurrent tumors at a median follow-up time of 21 months, while 4 of the 7 patients receiving incomplete resection suffered recurrence at a median follow-up time of 5 months. Multimodal therapy was given for the recurrent tumors and 4 of the seven patients showed no evidence of disease. Radiotherapy was given to 10 patients (the 7 recurrent tumors, 2 incomplete resections and the tumor with CSF seeding) and 5 of them recurred. The patient (No. 19) who was diagnosed with CSF seeding underwent radiotherapy after GTR, but died from the disease. The patient (No. 6) who underwent GKRS after STR was found to be stable after for 42 months. One patient (No. 11) received 2 cycles of temodal, but died.
Prognostic factors
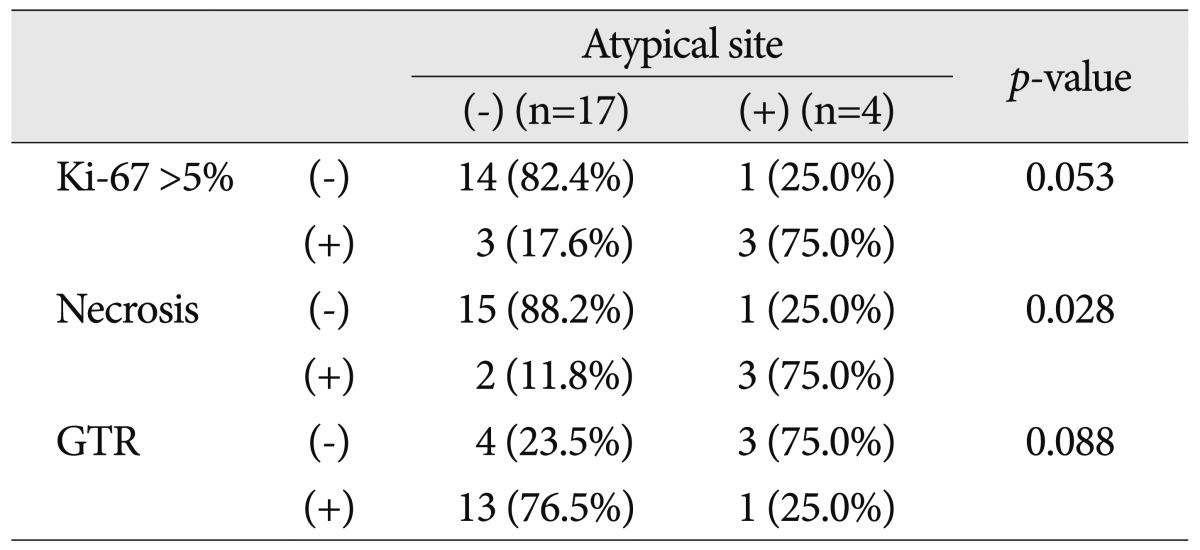
Table 4 shows the results of univariate analysis of prognostic factors for recurrence. Enhancement above moderate grade was not related to recurrence. However, peritumoral edema above moderate grade (p=0.022), atypical tumor location (p<0.001) and large tumor size (p=0.048) were correlated with recurrence. Ki-67 index, necrosis and GTR seemed to be related to recurrence rates, but their effects were not statistically significant. Atypical tumor location was the most important factor affecting prognosis. GTR was difficult to achieve when the tumors were at atypical sites. High Ki-67 value and necrosis were more frequent in tumors at atypical sites (Table 5).
DISCUSSION
PXA is a low-grade glioma (LGG) and overall survival is similar to other LGG. However, radiologic and pathologic findings of PXA suggest high-grade tumor. And some PXAs show rapid progression unlike other LGG5,8,23,35). Therefore, it is important to find prognostic factors and establish therapeutic modalities.
Clinico-radiological findings
The morphological features of PXA are clearly revealed by MR imaging4,19,33). The most common tumor location is the temporal lobe, followed by the frontal, parietal and occipital lobes. Involvement of the basal ganglia, cerebellum and spinal cord is rare2,6,8,25,28,36). Five tumors were located in an atypical site; two in the corpus callosum, one in the thalamus, one in the cerebellum and one in the lateral ventricle. One died of post-operative complications and three of 4 died of disease. Hamlat et al.8) noted that the clinicopathologic features of cerebellar PXA show some differences from PXA located in the cerebral hemispheres. Fu et al.5) reported intraventricular PXA with rapid progression. GTR was difficult to achieve at atypical sites and pathologic markers, including mitoses and necrosis, seemed to be related to poor prognosis (Table 5).
Recurrence occurred in five of the 9 patients with above moderate grade edema. Four patients who were diagnosed with PXA with anaplastic features displayed severe peritumoral edema. The mechanism of peritumoral edema is complex31). There is no consensus on how peritumoral edema should be measured. However, some studies reported that peritumoral edema is related with poor prognosis in high-grade gliomas9,27,29). Sheping et al. found that peritumoral edema was a sign of tumor infiltration but was not a prognostic factor36). However, Tien et al.33) concluded that it may point to a poor prognosis.
Tumor size as the prognostic factor for PXA was not well studied. However, it is known that large tumor volume is a negative prognostic factor in LGG1,26,30). Berger et al.1) suggested that the biology is different between small and large tumors. Smith et al.30) suggested that large tumors at presentation may grow faster than small tumors. Out of eleven patients with large tumors, GTR was not achieved in 6 patients (p=0.063) and 5 patients (p=0.149) showed high Ki-67 index. Five patients (p=0.035) with tumor necrosis were shown to be all large tumors. Surgical extent and biological difference of large tumors may influence the recurrence rate. In our study, atypical site, peritumoral edema and tumor size significantly affected prognosis. However, as the sample size was relatively low, further study is required.
Histopathological review
The cells in PXA are pleomorphic and often large, somewhat bipolar, with glassy cytoplasms and dark, lobulated nuclei reminiscent of malignant fibrous histiocytoma. However, the presence of GFAP identified the tumors as variants of astrocytomas12).
Mitotic index was the most significant predictor of recurrence and survival in a multivariate analysis performed by Giannini et al.6). They classified mitotic indices into three groups : 0, <5, and >5 mitoses per 10 high-power fields and suggested that the poor outcome group with >5 mitoses should be referred to as PXA with anaplastic features, although this remains controversial6,10). In our study, mitoses were seen in 12 patients, and most of those had mitotic indices of <2. Just 4 patients were diagnosed as suffering from PXAs with anaplastic features and their outcomes were diverse. Two of 3 patients suffering from PXAs with anaplastic features initially suffered rapid disease progression. However, the remaining patient and a patient with a tumor that became malignant remained free from recurrence. Pahapill et al.24) suggested that necrosis could be related to overall survival. Necrosis was present in 5 of our patients and was not correlated with disease progression. However, the four tumors with anaplastic features were necrotic. A larger data set might reveal a relationship between necrosis and disease progression. Usually the Ki-67 index in PXAs is very low (0-1%)4). In our study, six tumors were found to have Ki-67 indices above 5% and of those, 4 (67%) recurred (p=0.064). A biopsy of patient No. 11 showed no mitoses, a KI-67 index of less than 1% and no necrosis. Although the tumor was not malignant, the patient died at 15 months. Patient No. 20 had malignant biopsy results including 6 mitoses and a Ki-67 index of 12.6% with necrosis but was stable without adjuvant therapy for 21 months. There is a previous report of a patient with no mitoses, necrosis, or endothelial proliferation whose disease progressed, with spinal metastasis and malignant transformation to glioblastoma23). Since the pathologic factors determining prognosis are not well understood, it is difficult to identify anaplastic PXAs.
Treatment
Macaulay et al.21) reported no difference in overall survival between patients who had undergone GTR and those in which GTR was not achieved. However, many studies such as those of Pahapill et al.24) and Giannini et al.6) found that GTR conferred a significant overall survival advantage. Giannini et al.6) reported that extent of resection was the single most significant predictor of recurrence-free survival in a multivariate analysis. Although GTR was not a significant factor in our study because of the small sample, it may promote disease control and is the recommended treatment for patients with PXA like other glioma.
The findings regarding the use of adjuvant therapy such as radiation or chemotherapy are controversial. Giannini et al.6) did not specifically address this question in their report, because only a small number of patients received adjuvant radiotherapy. Pahapill et al.24) noted that the survival curves for those receiving adjuvant radiotherapy were not significantly different from those who did not. Therapeutic radiation effect was not apparent in Marton et al.'s22) report, either. Adjuvant therapy was not administered after GTR, even for PXAs with anaplastic features; in those cases the patients were followed up using MR. Radiotherapy seems to be ineffective, so far. However, Koga et al.15) reported that stereotactic irradiation was helpful for disease control of PXA as shown in the case (patient 6) of our study. The trial of various radiation therapeutic modalities is needed for treating recurrent tumors when GTR is not achieved and metastasis occurs.
The frequency of recurrence of PXA has been reported to be 30%, and that of malignant transformation 10-20%16,23,34). In our study, disease progression occurred in 7 patients (33%) similar to other reports, and malignant transformation occurred in one patient (5%). Marton et al.22) noted that surgery is also the therapy of choice for recurrent tumors without malignant transformation and radiotherapy and chemotherapy did not seem to play a significant role. Chemotherapy for PXA has been generally considered ineffective6,15,32). Fouladi et al.4) also noted that further surgery is recommended in the case of recurrent disease. Re-operation and GTR is the most important treatment for recurrent tumors; adjuvant therapy can be administered provided pathological markers and clinical progression are taken into account.
The last enrolled patient (No. 22) showed a typical presentation of PXA. His presenting symptom was seizures. The tumor morphology was a solid dominant, well-enhanced and well-demarcated cystic mass and was located in the left temporal lobe. A good prognosis was predicted as the patient showed low peritumoral edema and typical supratentorial cortical location on MR images, no mitoses, low Ki-67 index and no necrosis on the biopsy. After GTR was performed, adjuvant therapies were not administered. Adjuvant therapies can be administered to cases where recurrent tumors are shown to have metastasized or have aggressive extensions on MR images or new proliferative pathological markers are seen.
CONCLUSION
PXA shows good overall survival like other LGG. However, unlike a typical LGG, prognostic factors and therapeutic modalities are well not established. Peritumoral edema, atypical location and large tumor size were poor prognostic factors in our study. Although the difference between the effects of GTR and STR on prognosis was not significant, we suggest that GTR is the treatment of choice for primary and recurrent tumors. Adjuvant radiotherapy, especially stereotactic irradiation can be an option for recurrent, residual or metastatic tumors.

 XML Download
XML Download