

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemifacial spasm (HFS) is defined as unilateral, involuntary, irregular clonic or tonic movement of muscles innervated by the ipsilateral seventh cranial nerve21). More than three decades ago, Jannetta9,10) began to popularize microvascular decompression (MVD) of the seventh cranial nerve to treat HFS. Although the concept of neurovascular compression (NVC) and the rationale for MVD have been sufficiently clarified, there still are several questions that have not been adequately answered about its epidemiology and pathogenesis.
It is generally believed that HFS is caused by vascular compression of the root exit zone (REZ) of the facial nerve5,9-11). However, as has been proposed, pathophysiologic mechanisms other than vascular compression may contribute to the induction of HFS. HFS can appear as a sequela of peripheral facial nerve lesions, such as Bell's palsy or traumatic facial nerve damage, and most cases are sporadic10). Few familial variants of HFS have been described, but the nature of the inherited predisposition and its associated factors have been poorly understood until now. Carter et al.3) reported a pontine vertebral artery (VA) anomaly, and Miwa et al.13) detected atherosclerotic elongation of the VAs. In other studies, no vascular lesions were detected3,4,15,16) and genetic susceptibility in cranial rhizopathies was suggested2,13). Furthermore, progressive atherosclerotic vascular change, accelerated by aging and hypertension (HTN), was hypothesized as a determinant for the clinical appearance of HFS20).
In the present study, we collected comprehensive clinical data for our patients with familial HFS and compared them with sporadic HFS in order to identify predisposing factors for familial HFS and to explore the role of genetic susceptibility.
MATERIALS AND METHODS
Patient population and inclusion criteria
Thirty-nine HFS patients who had familial history had visited the department of neurosurgery at our medical institute from January 2001 to July 2011. The diagnosis of HFS was made by one board-certified neurosurgeon (K. Park) on the basis of characteristic clinical manifestations as well as neuroradiologic and electrophysiological findings.
Neuroradiologic studies were performed, including brain magnetic resonance imaging (MRI) and magnetic resonance angiography, and patients who had abnormal mass lesions other than blood vessels, which can contact or irritate the facial nerve, were excluded. Only one patient in the pedigree had visited our hospital was excluded to eliminate potential discrepancies in diagnosis and we could not evaluate their clinical findings. At least two family members diagnosed with HFS in our hospital were included in this study.
Therefore, our study included 20 patients and ten pedigrees. We investigated the following clinical characteristics in all patients : age at symptom onset, gender, side of spasm, past medical history such as HTN, the offending structures and characteristics on MRI and/or operative findings, surgical outcomes and the patients' pedigree. We interviewed patients via telephone to investigate their exact pedigree. We described each family, which spanned at least three generations, and indicated the occurrence of familial spasm in their pedigree. To compare the characteristics of familial HFS with those of sporadic HFS, we used data from patients who underwent MVD in our hospital between 1998 and 2010.
Neuroimaging evaluation
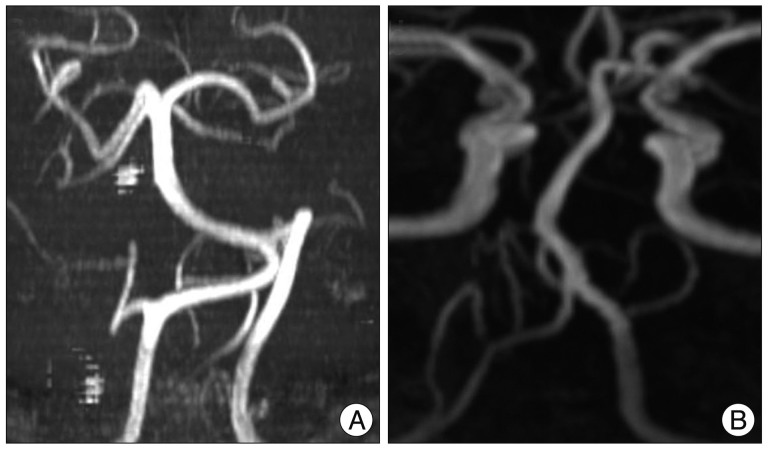
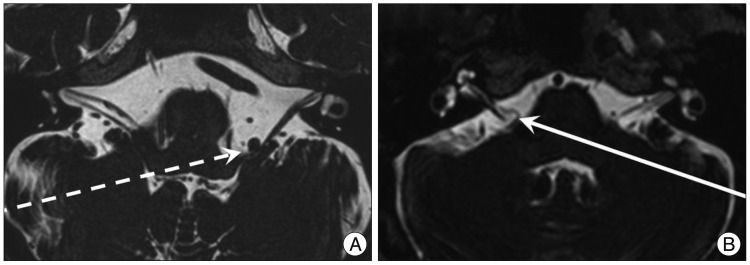
MRI scanning was performed using a GE Signa 1.5 Tesla scanner (GE Medical Systems, Milwaukee, WI, USA). The original MR angiographic slices were acquired in the transverse plane and reformatted using the maximum intensity projection technique. The morphology of the vertebrobasilar system was graded as follows : 0=normal; 1=tortuous but no direct contact with the REZ; 2=tortuous and direct contact with the REZ. Tortuous VA defined as an increase in the length and/or diameter of the VA enough to contact REZ of facial nerve (Fig. 1, 2). This retrospective study did not include MRI studied for other family member other than patients.
Surgery and surgical outcome
All of the included sporadic HFS patients and all but one patient with familial HFS underwent MVD at our hospital. All surgical procedures were performed via a retrosigmoid suboccipital approach with intraoperative monitoring, and this procedure has been well described in previous reports8,12,17,18).
All patients were followed-up postoperatively at the outpatient department at regular two- to three-month intervals. Evaluation and recording of the postoperative outcomes were carried out by an assisting nurse and the surgeon on postoperative days 1 and 3, at 3 weeks, at 3, 6 and 12 months, and more frequently as needed. A good outcome was defined as no residual spasm at the most recent follow-up. Minimum follow-up periods was 3 month.
Statistical analysis
The Statistical Package for the Social Sciences (SPSS version 18.0, SPSS Inc., Chicago, IL, USA) was used for data storage and statistical analysis. The data are presented as means±SDs. The chi-square test was used to assess the statistical significance of the independent variables between the two groups (familial HFS vs. sporadic HFS), and the Mann-Whitney test with Bonferroni's correction was used to compare the age of onset between the two groups. A p-value<0.05 was considered significant.
RESULTS
Sporadic hemifacial spasm
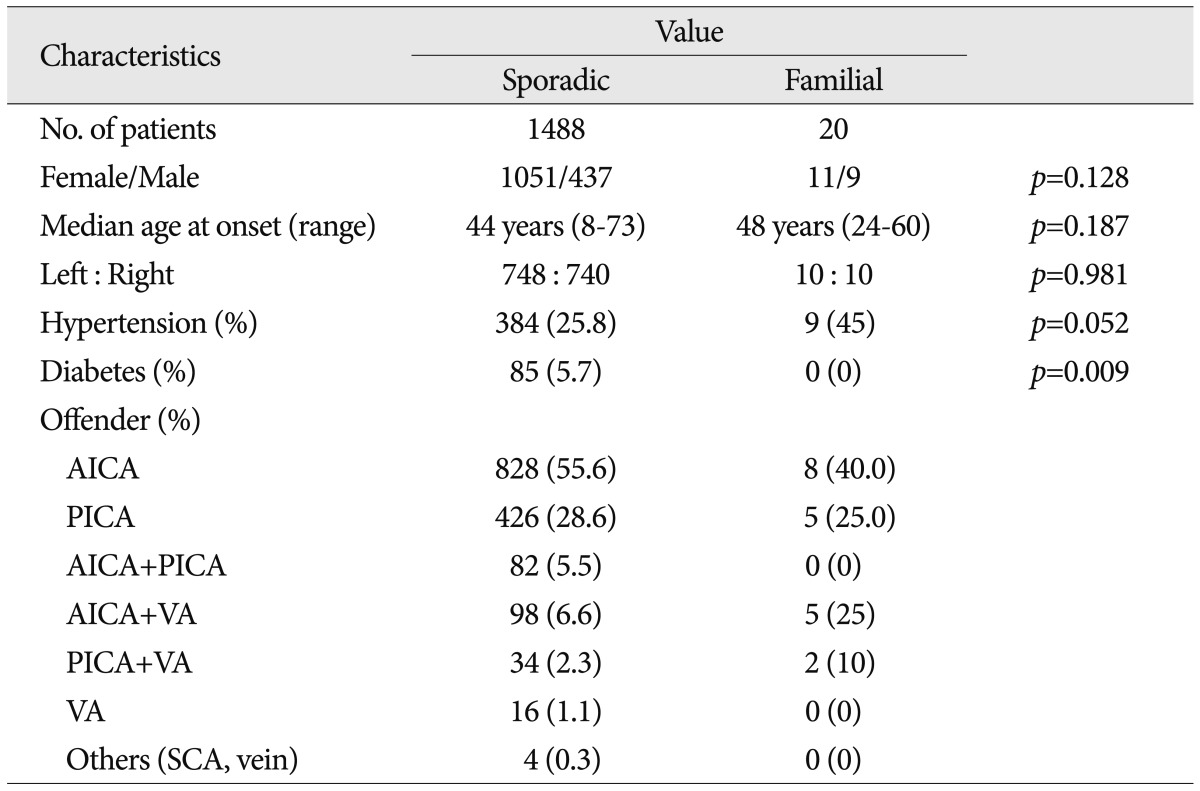
The 1488 patients who fulfilled the diagnostic criteria for HFS underwent MVD in our hospital. The mean age at onset was 40.7±10.7 years (mean±SD). We identified 748 patients with HFS on the left side and 740 patients with HFS on the right side. NVC of the ipsilateral facial nerve was caused by the anterior inferior cerebellar artery (AICA) in 55.6% of patients. Other offenders included the posterior inferior cerebellar artery (PICA) in 28.6%, the VA and AICA in 6.6%, the AICA and PICA in 5.5%, PICA and VA in 2.3%, and the VA alone in 1.1%. The frequency of HTN was 26.26%, and the prevalence of diabetes mellitus was 4.84% (Table 1). At the most recent follow-up examination, 1375 of the total 1488 (92.4%) patients were cured and 113 (7.6%) had residual or recurred spasm. We did not evaluate the tortuosity of VA in sporadic cases in this study because some imaging files were not available.
Familial hemifacial spasm
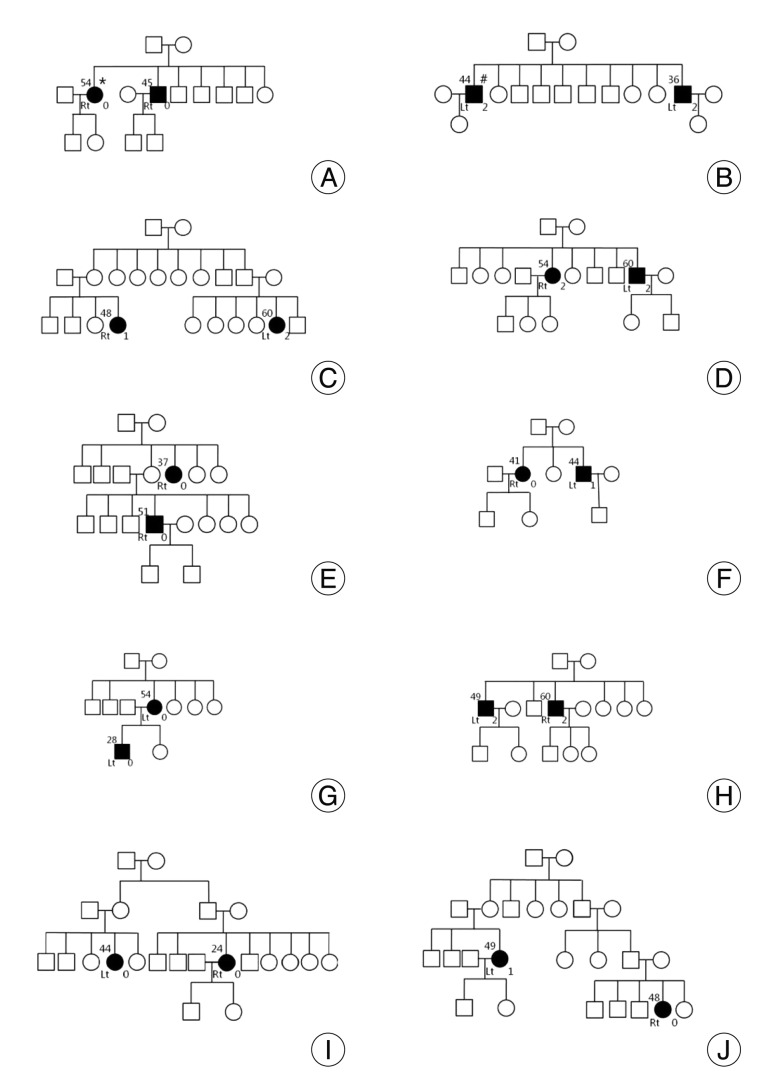
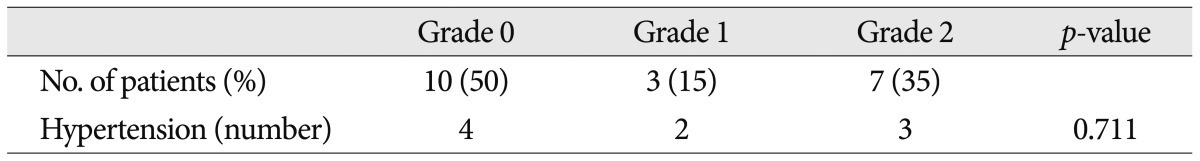
Twenty patients met the inclusion criteria of this study, and 19 of them underwent MVD at our hospital. Other 1 patient rejected the surgical treatment. The mean age at onset was 46.5±9.9 years. There was no significant difference of onset age between familial HFS and sporadic HFS (Mann-Whitney test, p=0.187). NVC by the AICA was found in 40% of familial patients. Other data are shown in Table 1 and Fig. 3 shows the patient pedigrees. According to the family trees depicted in Fig. 1, the inheritance pattern was difficult to define clearly using these data. Ten patients had HFS on the left side, while the other ten patients had it on the right side. A tortuous vertebrobasilar system was observed half of the familial HFS patients on preoperative MRI and/or intraoperatively (Table 2). Additionally, in seven out of ten, a tortuous VA was one of the offending arteries. In familial HFS patients, chi-square test revealed that the VA was a more frequent offender than in sporadic cases (35.0% vs. 10.0%, p<0.05). All patients in the B, C, D, and H groups had a tortuous VA (Fig. 1). One of the ten patients with tortuous VAs exhibited atherosclerotic changes on MRI.
The prevalence of HTN was higher in familial HFS patients than in sporadic cases, but the difference was not statistically significant (25.8% vs. 45%, p=0.052). Furthermore, the HTN did not correlate with the tortuous VA in familial HFS group (p=0.711) (Table 2). Eighteen out of 19 familial HFS patients who underwent MVD showed a good outcome. There was one case of facial palsy and one case of hearing loss. None of the patients had coexisting movement disorders or demyelinating diseases. Electrophysiologic studies, including pre and intra-operative electromyography show no difference between familial HFS and sporadic HFS.
DISCUSSION
Previous studies have suggested that irritation of the facial nerve due to its close contact with a blood vessel promotes hyperactivity and hyperexcitability of the facial nerve nucleus19). Until recently, factors that produced NVC or that caused predisposing conditions to progress to symptomatic NVC were poorly understood. We predicted that the analysis of familial HFS may help elucidate the pathogenesis of HFS.
Out of 21 patients with familial HFS that have been reported in the literature, 18 patients had symptoms on the left side3,4,6,15,16). Miwa et al.16) suggested that this lateralization may be related to the functional asymmetry of the supranuclear motor control of facial muscles. However, our data did not support this opinion. In this study, to the best of our knowledge the largest familial case series, the symptomatic side ratio was 1 : 1 (right : left) and was similar in patients with sporadic HFS. Furthermore, six out of ten pedigrees showed that each patient had contralateral sides affected in the same pedigree. Because of the paucity of cases, our findings cannot be generalized; however, genetic susceptibility might not be associated with specific side predilection.
Carter et al.3) reported that the age at onset in patients with familial HFS was below 30, suggesting that the age at onset of familial HFS is earlier than that of sporadic HFS. In contrast, another report described late-onset familial HFS13). In our study, two patients had early-onset HFS (<30 years old), but almost all of the remaining patients developed HFS after the age of 40. The age of symptom onset was quite different between HFS patients in the same pedigree (Fig. 1). When compared with sporadic HFS, we found no significant difference (p=0.187). According to our data, the age of symptom onset may not associated with familial HFS. However, we suspected that some aberration of genetic susceptibility could lead to early onset. Therefore, further studies are necessary to determine whether genetic factors influence the age of HFS onset.
Interestingly, we found that enlarged and tortuous (dolichoectatic) VAs were present in half of the familial HFS patients in our study compared to only 11% of sporadic HFS cases. In addition, the shape of the VA was similar to that of other patients in the same pedigree in eight separate families. Underlying morbidities, such as diabetes mellitus or HTN, did not correlate with VA tortuosity (p=0.653). In another series of 648 patients undergoing MVD, the cause of compression was the PICA in 68%, the AICA in 35%, and a dolichoectatic VA in 24%; other vessels have been involved in a minority of cases2). These data suggest that familial HFS had a higher rate of VA tortuosity and similar offenders compared to sporadic cases. Lagalla et al.13) hypothesized that tardive clinical manifestation of NVC could be due to progressive atherosclerotic vascular changes caused by the systolic pressure waves in chronic hypertensives. This hypothesis can explain the late onset of familial HFS associated with vascular tortuosity. But, we found no correlation between vascular tortuousness and HTN or diabetes, suggesting that genetic susceptibility may also play a role in the development of tortuous vessels and familial HFS.
There was no case of preoperative facial palsy, and all patients had a definite vessel on MRI compressing the facial nerve. The incidence of favorable outcomes was roughly the same in familial HFS patients who underwent MVD compared to sporadic HFS cases. This finding suggests that vascular compression contributes to the induction of mechanisms underlying not only sporadic cases but also familial HFS. Until recently, the role of direct contact between vessels and cranial nerves was not fully understood. According to the Lang14) findings, the anatomical course of the first part of the PICA forms a loop extending cranially that reaches as far as the lower border of the seventh and eighth cranial nerves in 15.5% of normal humans. This information suggests that NVC is not a single pathogenetic factor of HFS. Other genetic susceptibilities, such as increased excitability of the brainstem or vulnerability to minor cranial nerve damage, may play a key role in the development of HFS. Unfortunately, to the best of current knowledge, there is no known genetic factor associated with HFS7,22).
Some authors have suggested an autosomal dominant pattern of inheritance for HFS with/without low penetrance by identifying affected members in successive generations3,13,16). However, an inheritance pattern was difficult to define in our series. Nevertheless, autosomal dominant inheritance with partial penetrance could be hypothesized, yet we cannot exclude the possibility of an autosomal recessive inheritance pattern because we found only one pedigree that had patients with successive generations of HFS. Identified familial cases often showed different clinical features, thus we hypothesize that familial HFS may be a heterogeneous disease group with different genetic susceptibilities. Therefore, each genetic susceptibility can possibly be attributed to a different inheritance pattern. As a result, some cases of familial HFS cannot be clearly differentiated from sporadic cases1,3,13,16,22). Clarifying the role of genetic susceptibility in HFS through further study of each familial case may help improve understanding of the pathogenesis of this disease. Also, comprehensive clinical assessment including brain imaging of each family member in the pedigree may help determine the inheritance pattern.
Based on current theories of the pathophysiologic mechanisms as well as the results of this study, we suggest that the vascular tortuosity in vertebrobasilar system plays a crucial role in certain familial HFS cases. With the exception of this finding, the clinical features and treatment outcomes of familial HFS overlap with sporadic cases. Considering the general incidence of HFS and the fact that each study reported in the literature had distinct findings, familial HFS is likely not a chance phenomenon. We believe that familial HFS patients may be a heterogeneous group, and further study of familial HFS including clinical, anatomic, genetic and molecular information may help identify a gene or trait that can provide insight into the mechanisms of both idiopathic and familial HFS.
 XML Download
XML Download