

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Delayed cerebral ischemia (DCI) is still a significant risk factor for poor neurologic outcomes in patients with SAH [21]. Previous reports on the relationship with DCI mainly studied three mechanisms including cerebral vasospasm, cortical spreading depolarization, and microthrombosis [18]. Nevertheless, the exact mechanism is still unknown, and thus, efforts are ongoing to identify other mechanisms of the pathogenesis of DCI.
Autophagy refers to a homeostatic cellular process by the degradation of dysfunctional organelles and superfluous proteins [11,25]. In the SAH research area, the main focus of autophagy has been its possible association with early brain injury (EBI) in rodent models. Lee et al. [15] reported that autophagosomes and autolysosomes in neurons increased markedly from the first day of an SAH. In addition, the high conversion of microtubule-associated protein light chain-3 (LC3)-I to LC3-II and increased expression of BECN1 and cathepsin-D were observed. During the acute phase, rapamycin, which is an autophagy activator, decreased in brain edema, cortical apoptosis, and blood-brain barrier impairment compared to vehicle-treated SAH rats [26]. Mitophagy is selective autophagy that maintains health mitochondria by the selective removal of dysfunctional or damaged mitochondria [16,28]. Wu et al. [28] reported that the knockdown of phosphatase and tensin homolog-induced kinase 1 (PINK1) and parkin was associated with the intracellular accumulation of mitochondrial fragments and damaged mitochondria via reduced mitophagy in endothelial cells. Cao et al. [1] also showed the neuroprotective effect of inhibiting mitophagy-associated nod-like receptor protein 3 inflammasomes against EBI after SAH. Compared to EBI, few studies have investigated the possible role of mitophagy in the development of DCI. Chou et al. [4] reported that higher mitochondrial membrane potential was associated with favorable functional outcomes in SAH patients. Youn et al. [30] reported that DCI patients had significantly decreased mitochondria membrane potential in CSF cells than non-DCI patients. These results suggest a possible association of mitochondrial dysfunction with DCI pathogenesis. A follow-up investigation showed that mitochondrial dysfunction associated with autophagy and mitophagy might have a role in DCI development [31]. The DCI patients showed a higher expression of death-associated protein kinase (DAPK)-1, BCL2 interacting protein 3 like (BNIP3L), and PINK1 than the non-DCI patients. In particular, DAPK1 was the most significant one among markers differentially expressed between those with DCI and those without DCI.
Bioinformatics analysis has been increasingly reported in various medical conditions in anticipation of suggesting new insight into the disease pathogenesis. An epigenome-wide association study found that hypermethylation of the INSR gene (cg00441765) and the CDHR5 gene (cg11464053) was closely related to DCI [13]. This result indicated a simple difference in the methylation frequencies of CpG sites according to DCI. Subsequent bioinformatics analysis revealed additional information on various biological processes (BP) and hub genes associated with DCI, which existing analysis methods do not provide. Here, using bioinformatics analysis of the raw data, we further evaluated the interactions among the differentially expressed autophagy and mitophagy biomarkers of CSF cells associated with DCI.
MATERIALS AND METHODS
This study was approved by the Institutional Review Board (No. 2017-9, 2018-6, and 2019-6) of the hospital and informed consent was obtained from the patients or their relatives.
Data acquisition and process
The original dataset was derived from an ongoing study entitled “The First Korean Stroke Genetics Association Research” of the five university hospitals that prospectively collected the genome and protein databank of patients with various cerebrovascular diseases since March 2015 [12,17]. In this databank, the CSF samples of SAH patients were investigated to determine whether mitochondrial dysfunction associated with autophagy and mitophagy may be related to DCI following SAH (Fig. 1). The expression of the markers was measured using real-time reverse transcription-polymerase chain reaction (qRT-PCR) and Western blotting. The markers measured by qRT-PCR were 1) DAPK-1; 2) BNIP3L; 3) Bcl-1 antagonist X (BAX); 4) PINK1; 5) Unc-51 like autophagy activating kinase 1 (ULK1); and 6) nuclear dot protein 52 (NDP52). The markers measured by Western blotting were 1) BECN1 (autophagy executor gene); and 2) p62 (autophagy adaptor protein) [5,24,31].
Bioinformatics analyses
Gene ontology (GO) enrichment (https://tools.dice-database.org/GOnet) and Kyoto Encyclopedia of Genes and Genomes pathway (https://www.genome.jp/kegg/mapper/color.html) analyses were carried out using the visualization tools [32]. GO terms with a p-value of less than 0.05 were defined as significantly enriched. Protein-protein interaction (PPI) network analysis was performed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING). The minimum required interaction score was set to the highest confidence of 0.7 [19,23].
RESULTS
Identification of autophagy and mitophagy markers
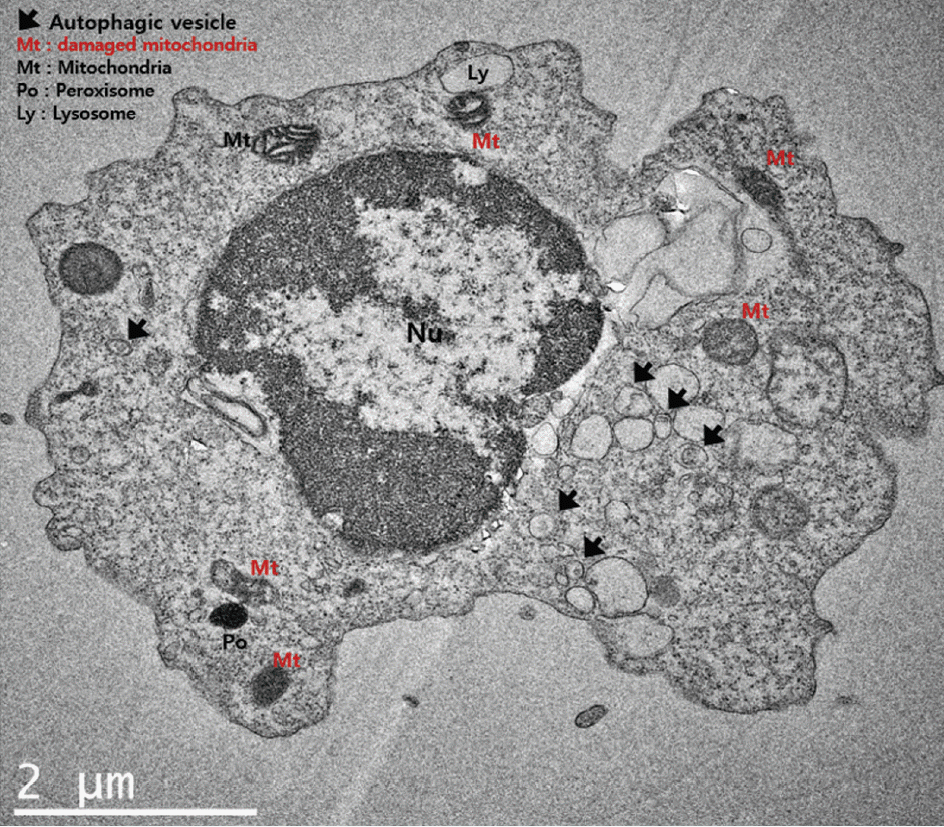
Data on CSF cells obtained from 56 SAH patients were selected for the analysis. Among them, DCI was observed in 22 patients (38.6%). A flow chart of the study is presented in Supplementary Fig. 1. Transmission electron microscopy showed various mitochondrial conditions in DCI such as damaged mitochondria and disarrayed cristae with autophagic vacuoles and lysosomes (Fig. 1). Among the various markers, DCI patients exhibited significantly increased mRNA levels of DAPK1, BNIP3L, and PINK1, and protein expression of BECN1 with p62 degradation than compared to the non-DCI patients.
In silico functional analysis
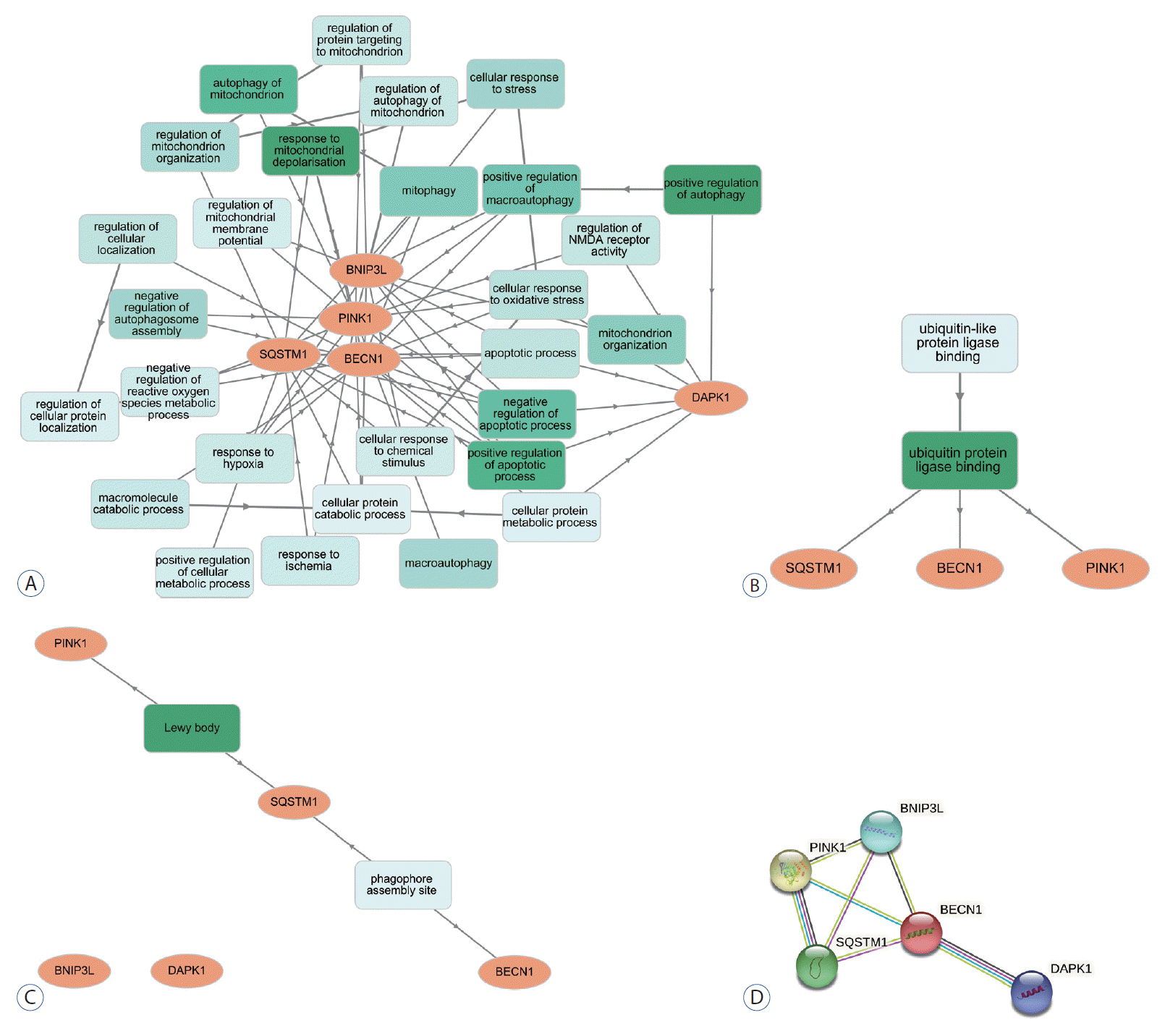
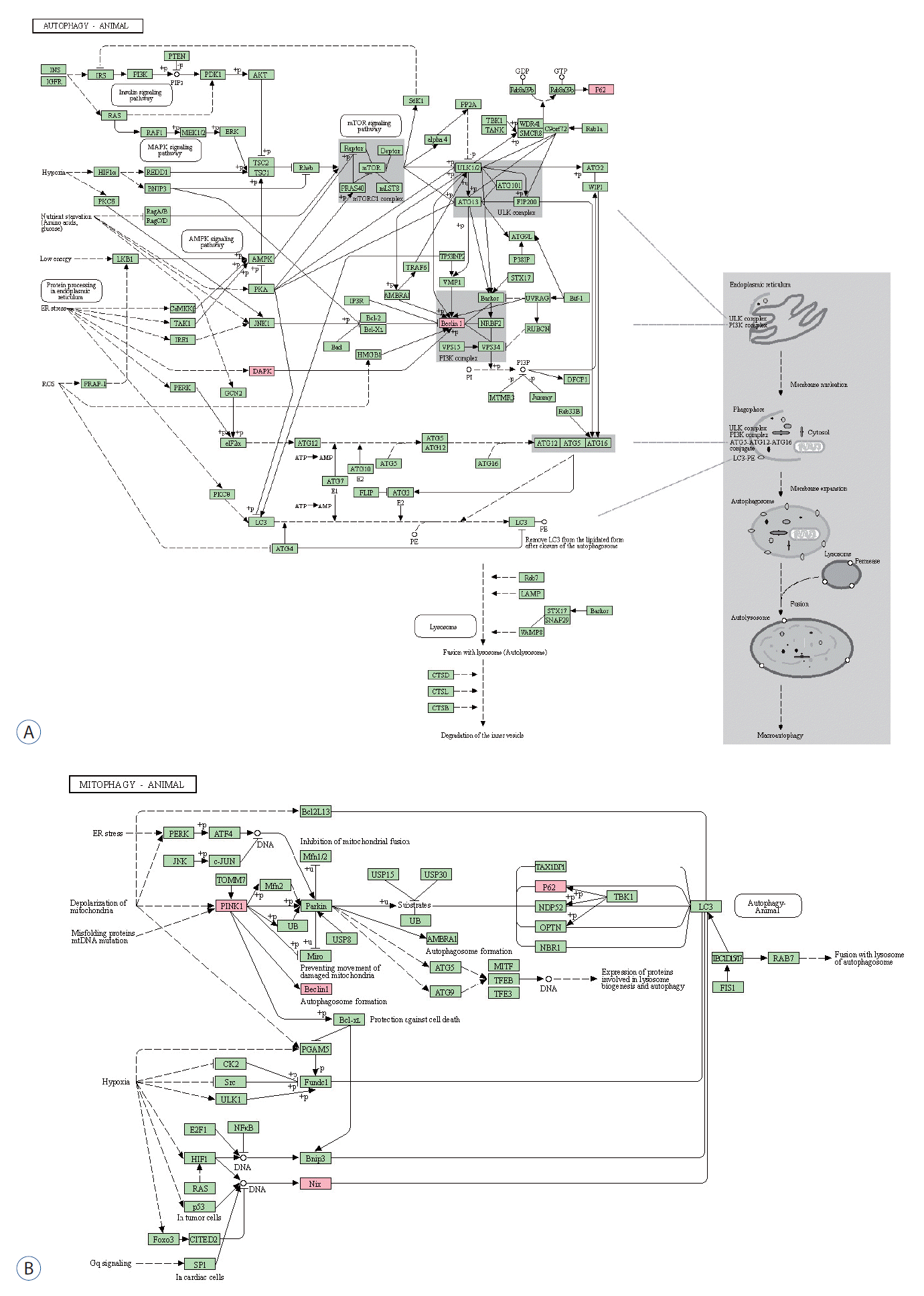
GO enrichment analysis was performed for BP, molecular function (MF), and cellular components (CC). The most enriched BP functions were the positive regulation of autophagy, followed by the response to mitochondrial depolarization, the positive regulation of apoptosis, and mitochondrial autophagy in the development of DCI (Table 1 and Fig. 2A). In MF, DCI was associated with ubiquitin-like protein ligase binding and ubiquitin-protein ligase binding (Fig. 2B). In the cluster of CCs, Lewy bodies and the phagophore assembly site were the most enriched in DCI patients (Fig. 2C). Three and four of the differentially expressed genes are marked in red in the autophagy and mitophagy pathways in DCI development in Fig. 3, respectively. PPI analysis revealed that BECN1 was the most connected gene among the differentially expressed markers associated with autophagy and mitophagy in SAH patients with DCI (Fig. 2D).
DISCUSSION
Few studies have investigated mitophagy associated with DCI in SAH patients, although mitophagy research has been actively conducted in other brain diseases. Conventional mitophagy is regulated by the PINK1/parkin pathway [6]. In the damaged mitochondria of patients with autosomal recessive Parkinson’s disease, PINK1 is expressed on the outer membrane during activated E3 ubiquitin ligase activity, which recruits parkin. This selective lysosomal removal was followed by parkin-mediated polyubiquitination [6,20]. Parkin-mediated mitophagy was also involved in the cardiac stress-response mechanism [7]. Kubli et al. [14] reported that parkin-deficient mice showed increased mitochondrial dysfunction associated with decreased mitophagy myocardial infarction. However, alternative parkin-independent mitophagy or autophagy adapter proteins are also involved in mitophagy [6]. Phosphorylated Unc-51-like kinase 1, which interacts with Ras-related protein 9 at serine 179 followed by complex formation with Rip 1 and dynamic-related protein (Drp) 1 were observed in the heart in ischemic and glucose-deprived states. After phosphorylated Drp1 was sequestrated, the lysosomal degradation of mitochondria was observed [6,22]. In SAH patients, a higher expression of PINK1 seemed to be associated with DCI. And DAPK1 and BNIP3L were significantly higher in DCI patients than in non-DCI patients. Our additional bioinformatics analysis showed that the positive regulation of autophagy was mostly enriched, followed by the response to mitochondrial depolarization in BP in DCI development. DAPK1, BNIP3L, BECN1, and PINK1 were involved in the positive regulation of autophagy, and BECN1, PINK1, and p62 in the response to mitochondrial depolarization. PPI analysis also demonstrated that BECN1 was the most connected gene among the highly expressed biomarkers in DCI development. BECN1 is expressed within cytoplasmic structures such as mitochondria and the endoplasmic reticulum and contains three domains including the Bcl-2 homology motif, the coiled-coil domain, and the evolutionarily conserved domain (ECD) [8]. BECN1 interacted with PI3KCIII/Vps34 via ECD and positively regulated the autophagy reaction [8]. Diabetic mice showed increased autophagosome formation and autophagic flux with a higher expression of BECN1, microtubule-associated protein LC3-II, and a lower expression of p62 [27]. BECN1 has been suggested as a cancer target for anti-tumor agents that enhance BECN1 activity. Based on these findings, it is necessary to investigate which specific mechanisms and proteins can be therapeutic targets for DCI in the future.
Several clinical variables such as hypertension and sex differences may affect autophagy and mitophagy in DCI. In DCI patients, blood pressure was maintained at high levels to increase blood flow to the brain. Hypertension itself may affect autophagy and mitophagy, although the exact mechanism remains undetermined. Increased BNIP3 expression was correlated with pressure overload in heart failure [2]. Givvimani et al. [10] reported that mitophagy inhibitor ameliorated heart failure by decreasing pressure overload concomitant with the decreased expression of LC3 and p62. Gender can influence not only DCI occurrence but also autophagy and the mitophagy process. The rate of DCI was significantly higher in females compared to males [9]. Chen et al. [3] reported that female rats exhibited a higher ratio of LC3B to LC3A in a cardiac ischemia-reperfusion model. Regarding the two pro-apoptotic proteins of phosphor-p38 and Bax, male rats showed higher levels of phosphor-p38, and female rats showed lower levels of Bax after ischemic-reperfusion. Female ovariectomized rats exhibited an increased expression of LC3 and BECN1 with decreased p62 [29], suggesting that autophagy and mitophagy may vary depending upon sex hormones. Although, there were no significant differences in hypertension and gender between the DCI and non-DCI groups in the original dataset, additional bioinformatics analysis adjusting for possible clinical variables that may affect the results is required in a large SAH population.
This study had some limitations. First, the relatively small number of enrolled patients in the original data is a concern, although this was the first bioinformatics analysis of autophagy and mitophagy markers related to DCI. Second, we used some representative biomarkers of autophagy and mitophagy. Thus, in addition to the biomarkers we studied, follow-up studies are needed, including many biomarkers to get more accurate results. Third, bioinformatics results are largely dependent upon the quality of the raw data. To address this issue, it is necessary to perform the study considering various medical conditions that may affect the outcomes.
CONCLUSION
We comprehensively analyzed autophagy and mitophagy markers associated with DCI using bioinformatics analysis. These attempts can be helpful for understating DCI pathogenesis. We also identified a hub gene of BECN1, which is expected to be a therapeutic target for future DCI treatment in patients with SAH.
 XML Download
XML Download