

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Caudal agenesis refers to the spectrum of disease with the common component of ‘agenesis of the bony spine of the lower sacral and coccygeal region’. Despite its simplified nomenclature, it may involve varying degrees of anomalies of the lumbosacral spinal cord and be part of complex syndromes of numerous organs. Understanding the wide range of symptoms and related conditions based on the pathoembryogenesis will guide clinicians in the management of these patients.
Go to : 
DEFINITION
Caudal agenesis is a congenital anomaly involving the lower sacral and coccygeal spinal segments. Sacral agenesis and caudal regression have been used synonymously. The definition is based on agenesis of the spinal bone, but abnormalities in the spinal cord and a wide variety of complex anomalies are found. Associated anomalies develop in urogenital, anorectal, respiratory, and cardiac areas [2,5,11]. Because the disease was defined on the basis of the bony anomaly, its name is only a partial description. The involved bone is always missing (hence the term ‘agenesis’), but neural elements may be ‘not formed’ or ‘not degenerated’. In the context of secondary neurulation, these failures are explained as ‘failure of formation’ and ‘failure of regression’ (Fig. 1) [8].
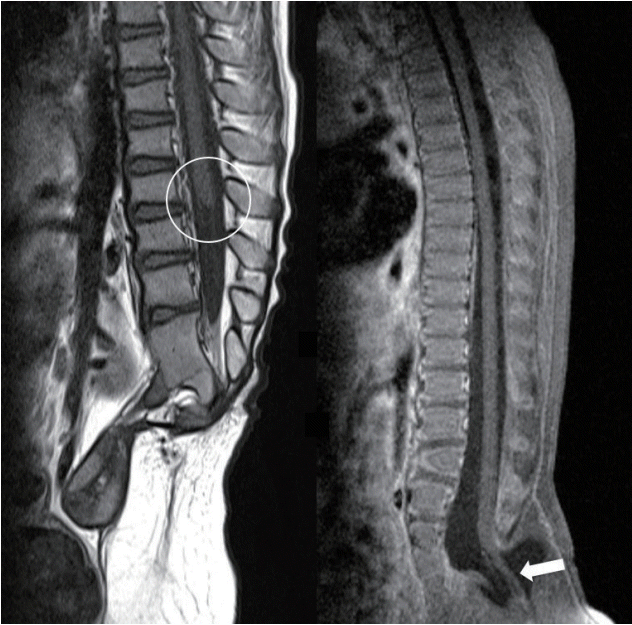 | Fig. 1.Left : Failure of formation type of caudal agenesis shows the blunt ended conus (circle) usually at the location at or above L1. Right : Failure of regression type of caudal agenesis shows low-lying conus. This case shows the retained medullary cord with the conus below the upper sacral level (arrow).
|
Because complex anomalies of different systems are frequently associated, caudal agenesis is a component of various syndromes, including VACTERL (vertebral anomaly, anal atresia, cardiac anomaly, tracheoesophageal fistula, renal anomaly, limb anomaly), OEIS (omphalocele, cloacal exstrophy, imperforate anus, spinal defects), and the Currarino triad (caudal agenesis, presacral mass, anorectal anomalies).
Go to :
PATHOEMBRYOGENESIS
The wide spectrum of anomalies (failure of formation vs. regression, association with multiple anomalies) may be explained by the complex embryological process of secondary neurulation. The caudal cell mass (CCM) is an undifferentiated cell mass in the area of the primitive streak, and it is the main player in secondary neurulation because the secondary neural tube is derived from it. However, the CCM is not only involved in the formation of the spinal cord and the vertebral bodies in the lower sacral and below, it is also closely related to the formation of surrounding structures [10], such as the genitourinary tract and anorectal organs. The hypoplasia and decreased ventral and caudal push of caudal mesenchymal tissue lead to the anomalous anteroposterior septation of the cloaca and defective closure of abdominal wall and cloacal membrane area. Weak ventral push of the caudal mesenchyme makes posterocaudal deviation of the urorectal septum (which is an inward growth of caudal mesenchymal tissue from the both lateral walls of cloaca) causing rectal stenosis and imperforate anus. Deficient ventral movement of lateral body fold in the abdominal wall closure and rupture of the enlarged cloacal membrane may bring about gastroschisis, omphalocele, exstrophy of cloaca or bladder, epispadia, hypospadia and bifid scrotum. Caudal mesenchymal hypoplasia may affect the formation and migration of the kidneys leading to renal agenesis, and horseshoe or low-lying kidneys. Detailed pathoembryogenetic theories on caudal mesenchymal hypoplasia are available in our previous publication with schematic drawings [8]. We also suspect that the malformation and malfunction of the distal bowel may affect formation of esophagus and proximal bowel (tracheoesophageal fistula and omphalocele). The relationship between the heart anomalies and the disordered late gastulation at the area of Hensen’s node and the rostral part of primitive streak was discussed in another chapter in this Pediatric Issue, ‘Disorders of secondary neurulation’. However, it also seems possible that certain molecular abnormalities or other primary events may underlie the multiorgan involvement.
For specific lesions of the spinal cord, anomalies from earlier periods create the ‘failure of formation’ type, and anomalies in later periods result in the ‘failure of regression’ type.
Go to :
ETIOLOGY
It is difficult to know the true incidence of caudal agenesis because of its wide clinical spectrum. Patients with minimal involvement may not have symptoms, whereas patients with severe complex anomalies may not survive until birth [12]. Even after birth, approximately one-fifth of the patients are not diagnosed until the age of 3 or 4 [7]. The incidence is reported to be 0.01 to 0.05 per 1000 live births [1].
Most cases seem sporadic, and there may not be a strong genetic background for the anomaly. Familial occurrence was reported in a few cases, which suggests the possibility of autosomal recessive inheritance. For Currarino syndrome, the association with the mutation of the transcription factor MNX1 has been found in familial cases [2]. An animal model with mutation of the Brachyury gene in the caudal notochord showed the phenotypes of caudal agenesis, including skeletal and urorectal anomalies. However, no firm genetic association has been found in sporadic cases of caudal agenesis.
Maternal diabetes is the most commonly associated environmental factor (16–40% of caudal agenesis patients) [12]. One out of 350 babies of diabetic mothers have caudal agenesis [3,9], and there is a 200-fold increase in the incidence of caudal agenesis in mothers with diabetes. Other factors, such as longitudinal kinking of the fetus and exposure to insulin or retinoic acid, were suggested.
Go to :
CLASSIFICATION
The classifications of caudal agenesis found in the literature are based on bony anomalies [2,6,12]. The most commonly used system, Pang’s classification, identifies the complete absence of sacrum with or without lumbar vertebrae agenesis as types I and II, respectively. S1 is present in type III, but the lower sacral segments are missing to varying degrees. Type IV consists of various forms of hemisacrum. Type V includes coccygeal agenesis cases (Table 1). Although classification according to the bony anomaly seems intuitive, it may not represent the extent of spinal cord anomalies. We further integrated the concept of “failure of formation/regression” in the classification of caudal agenesis cases by classifying the cases based on the level and shape of the conus (Fig. 2). Analysis of our series of 74 patients revealed that classification by the level and shape of the conus was significantly more associated with the degree of neurological deficits than classification based on the bony anomaly type or the simple classification by level of the conus (above L1 vs. below L1) (unpublished data).
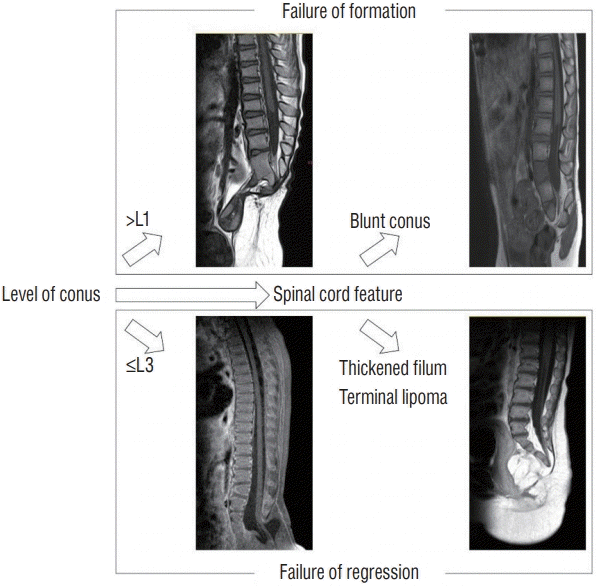 | Fig. 2.A schematic drawing summarizing a new classification system integrating the level of conus and the feature (shape and associated anomaly) of the spinal cord. This classification allows the designation of cases according to the pathoembryogenesis (failure of formation vs. failure of regression).
|
Table 1.
Classification of caudal agenesis based on bony anomaly
| Type | Subtype | Description | Configuration* |
|---|---|---|---|
| I | Total CA; some lumbar vertebrae also missing | ||
| IW† | Ilia articulate with sides of the lowest vertebra, maintain relatively normal transverse pelvic diameter |

|
|
| IN† | Ilia articulate or fused with each other below last vertebra, severely shortening transverse pelvic diameter |

|
|
| II | Total CA; lumbar vertebrae not involved | ||
| IIW† | Ilia articulate with sides of the L5 vertebra, maintain relatively normal transverse pelvic diameter |

|
|
| IIN† | Ilia articulate or fused with each other below L5 vertebra, severely shortening transverse pelvic diameter |
|
|
| III | Subtotal CA; at least S1 is present, sacrum lacks four, three, two, or one of its caudal segments. Ilia articulate with side of rudimentary sacrum, maintain normal transverse pelvic diameter |

|
|
| IV | Hemisacrum | ||
| IVA | Total hemisacrum; all sacral segments present on one side, but entire opposite side is missing‡ |

|
|
| IVB | Subtotal hemisacrum, unilateral; all sacral segments present on one side, only part of opposite side is missing |

|
|
| IVC | Subtotal hemisacrum, bilateral; part of each side is missing but to different extent |

|
|
| V | Coccygeal agenesis | ||
| VA | Total |

|
|
| VB | Subtotal |
Reprinted and modified from Lee et al. [8] with permission from Springer. *The small circles represent sacral foramina; total of four when five sacral pieces are present; presence of one foramen means two sacral pieces are present, etc. Flanking structures represent iliovertebral articulations.
![]()
Go to :
CLINICAL MANIFESTATION
Patients with caudal agenesis may present as part of a complex syndrome involving multiple systems or in association with genitourinary or anorectal anomaly [12]. Caudal agenesis is also diagnosed in patients with orthopedic conditions, such as dysplasias of the lower extremity or narrow pelvis. Patients may also present with progressive neurological symptoms suggestive of cord tethering. Caudal agenesis is also found incidentally on plain spine radiographs obtained for irrelevant reasons.
Physical examination may reveal a flat buttock, narrow profile of the pelvis, marked loss of muscle bulk in the calves, and scoliosis according to the level and degree of bony and neural anomalies. Neurological deficits, including motor/sensory/bladder/bowel/sexual dysfunctions, are frequently found in caudal agenesis patients. A discrepancy between the level of the motor deficit and the sensory deficit is consistently seen in these patients. Sensory functions are generally ‘spared’ several levels more than motor deficits. The reason for this ‘sparing’ is not known, but the dorsal root ganglia may separate from the neural tube in very early stages of development, which avoids the insult that causes caudal agenesis. Differences in the blood supply between the ventral and dorsal spinal cord and preservation of the dorsal conus compared to the ventral conus (dorsally elongated tip of conus) were also suggested [4].
Go to :
DIAGNOSTIC WORKUP
Plain spine radiography should be performed to document the presence and degree of the bony agenesis. Computed tomography may be performed in cases where vertebral elements are severely deformed and misaligned. Spine magnetic resonance imaging (MRI) is important to study the level and shape of the conus and dural sac. MRI reveals the presence of associated tethering lesions, such as a thick filum terminale, lumbosacral lipomatous malformation, retained medullary cord, terminal myelocystocele, and the Currarino triad.
Go to :
TREATMENT
Early intervention in caudal agenesis patients is important for the treatment of associated anomalies in pulmonary, gastrointestinal, or urorectal problems, including tracheoesophageal fistula, cloacal anomaly, omphalocele, bladder exstrophy, and imperforate anus.
Neurosurgical management is generally less urgent, and it is indicated primarily for patients with the ‘failure of regression’ type. Although the ‘failure of formation’ group may have neurological manifestations, the symptoms are static in nature and unlikely to benefit from untethering procedures. The ‘failure of regression’ group may suffer from progressive neurological deficits due to the tethering caused by spinal cord lesions. Dural or bony stenosis may also cause neurological deficits or pain in rare cases for whom decompression should be performed [12].
Go to :
 XML Download
XML Download