

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Sacrococcygeal teratoma (SCT) is a rare neoplasm of early infancy. SCT is a congenital tumor usually diagnosed in utero or during the neonatal period. Histologically, the majority of SCTs are mature teratomas, one of the subtypes of germ cell tumors (GCTs). Despite its histological similarity, SCT is different from GCTs developing in the gonads and other extragonadal sites after puberty. SCT is a benign neoplasm, and surgery provides a cure for the disease. However, recurrence and malignant transformation into teratocarcinoma or yolk sac tumor (YST) is possible. Patients with SCT are also commonly plagued with many gastrointestinal, urological, and neurological abnormalities. Despite the huge impact of this tumor on the quality of life of affected children, many of clinically important questions on SCT are currently difficult to answer. The major difficulties of clinicians dealing with SCT are the extremely low incidence of the disease and the lack of clear, unified guidelines for its management. Regarding the pathogenesis, the occurrence of SCT in such an unusual location at such a young age has been an enigma for a long time. Discoveries in developmental biology and molecular genetics have revealed the origin and pathogenesis of SCT to a great degree. This review will focus on the current knowledge of the clinical aspects and biology of SCT from developmental perspectives.
Go to : 
EPIDEMIOLOGY
Approximately 90% of human GCTs develop in the gonads, especially in the testis, but a few cases develop in extragonadal sites such as the brain, neck, mediastinum, and retroperitoneum. However, in neonates and young infants, the sacrococcygeal area is the most common site for GCT development. SCT develops from the sacrum and coccyx, protruding outwards and growing into the pelvic cavity. It is a rare disease with an incidence of 1 in 20000–40000 live births [4]. There is a clear female predominance with a 3 : 1 to 4 : 1 female-to-male ratio [35,39], which is in contrast to the well-recognized male preponderance of adolescent GCTs (both gonadal and extragonadal). Testicular GCTs (TGCTs) have one of the highest familial risks among cancers : having a brother with TGCTs increases the risk by 8–10 times and its presence in dizygotic or monozygotic twins elevates the risk by 37- or 76.5-fold [16]. Interestingly, most patients with SCT are sporadic, and no definite familial tendency exists, although a family history of twin gestation is observed in 10–15% of patients [4].
In a thorough examination, associated congenital anomalies are found in 15–30% of patients with SCT [17]. The most common anomalies are in the urogenital system, such as hydronephrosis, which can be considered as a consequence of compression by the tumor mass. Although the proportion of congenital anomalies is higher than that of the normal population (3–4%), causative associations are rare except for the Currarino triad. The Currarino triad is a very rare syndrome and consists of sacral bony defects, anorectal anomalies, and a presacral mass [15]. Approximately 40% of the presacral masses in the Currarino triad are benign teratomas [15]. The Currarino triad is an autosomal dominant syndrome linked to mutations in the HLXB9 gene [21].
GCTs are characterized by dramatic geographical differences in incidence according to the tumor location. The incidence of TGCTs in Europe is approximately 10 times that in East Asia [8]. Conversely, the incidence of intracranial GCT is five times higher in East Asia than in western countries [32]. Currently, there is no evidence of such a geographical difference of in the incidence of SCT. However, a nationwide study from Finland showed a higher birth prevalence of SCT (1 in 10000), supporting some genetic contribution to the etiology of SCT [31].
Go to :
PATHOGENESIS
The pathogenesis of SCT should be understood in the broad context of GCT development. All GCTs have been thought to be derived from primitive germ cells (germ cell hypothesis) [42]. The hypothesis explains the peculiar anatomical distribution of extragonadal GCTs along the midline of the body [28]. Primordial germ cells (PGCs) migrate from the yolk sac to their final destinations in the gonadal ridges by passing the hindgut where SCT can arise [22]. However, research on embryos and embryonic stem (ES) cells revealed that teratomas could be derived from ES cells, making ES cells candidate cells for developing into SCT [6]. Stem cells residing in extraembryonic tissues can also induce teratoma formation, adding other candidate cells to the possible origins of SCT [41]. The sacrococcygeal region also corresponds to the primitive streak and Hensen’s node in early embryonic development. If there are some residual totipotent stem cells from Hensen’s node, they can develop later into teratoma or YST, the two most common pathologies encountered in SCT [13,14].
Genomic sequencing of gonadal and extragonadal GCTs revealed that the majority of GCTs lack driver mutations for cancer initiation and progression. TGCTs are bona fide malignancies, but they are among the human cancers with the least mutational burden (0.3–0.5 mutation/Mb) [19,40]. TGCTs are divided into seminomas (germinomas in extragonadal sites) and nonseminomas, including embryonal carcinoma (EC), YST, choriocarcinoma, and teratoma. Mutations in KIT and KIT signaling (KRAS, NRAS) are found in 20–60% of seminomas, but fewer mutations are observed in nonseminomas [19,40]. TP53 mutations, a strong common driver of human cancers, are virtually not found in TGCTs. These findings indicate that GCT is not a mutation-driven tumor but a development-driven neoplasm.
Oosterhuis and Looijenga [27] proposed a unifying classification scheme for all GCTs of the human body. They envisaged GCTs as tumors derived through genomic reprogramming from their developmental cells of origin [27]. In their scheme, there are five types of GCTs [28]. Type I GCTs are infantile GCTs arising exclusively from extragonadal sites, with the sacrococcygeal region as the most frequent location. Type I GCTs are characterized by a female preponderance and consist of teratomas and YSTs. Rarely, somatic malignancy develops from mature teratoma through transformation [48]. Type I GCTs are believed to be derived from early PGCs during their migration. Migrating PGCs depends on KIT- KITLG (KIT-ligand) signaling for their survival. If early PGCs are reprogrammed to attain pluripotency by an abnormal microenvironment and to become ES cells, they can survive without the KIT pathway and form teratomas. A genome-wide association study (GWAS) revealed that a variant of the anti-apoptosis gene BAK1 is associated with type I GCTs, suggesting that evading apoptotic death after the loss of KIT signaling is important for type I GCT pathogenesis [33].
Type II GCTs are gonadal and extragonadal GCTs in adolescents and young adults [28]. If late PGCs in gonads and in extragonadal sites (ectopically located PGCs) are reprogrammed into totipotent ES cells, they develop into seminoma (germinoma/dysgerminoma) as a default pathway, and further neoplastic transformation leads to EC [27]. Through totipotent differentiation of EC cells, choriocarcinoma, YST, teratoma, and mixed GCT can subsequently develop. Type III GCTs are spermatocytic tumors (former spermatocytic seminomas) arising in elderly. Type IV GCTs are a dermoid cyst of ovary (cystic mature teratoma of ovary), which is thought to develop through parthenogenetic activation of the primary oocyte. Type V GCTs are a hydatidiform mole resulting from the fertilization of two sperm with an empty ovum (male parthenogenesis). Oosterhuis and Looijenga later added type 0 GCTs (conjoined twins) and type VI GCTs (reprogramming of somatic cells or induced pluripotent stem cells) to their scheme [20].
Studies on genomic imprinting in GCTs support this conjecture of developmental cell origin. Zygotes maintain biparental imprinting patterns during somatic development. However, in germline development, migrating PGCs start to lose their inherited imprinting patterns (partially erased), and upon arriving in the gonadal niche, all imprinting patterns are completely erased in late PGCs. The paternal and maternal imprinting patterns are re-established through germ cell development into mature spermatocytes and oocytes [25]. As expected, type I GCTs have partially erased genomic imprinting, reminiscent of early PGCs, and imprinting patterns are completely erased in type II GCTs which develop from late PGCs through reprogramming [28].
Therefore, the germ cell hypothesis is correct because all GCTs (type I to type V) develop from germ cells of different developmental statuses. Genetic polymorphisms, gains of genetic mutations, and epigenetic (or microenvironmental) changes affect the fate of germ cells and induce reprogramming to ES-like cells that lead to GTCs.
Go to :
CLINICAL FEATURES
Approximately 25–50% of SCTs are diagnosed in utero before birth (i.e., true congenital neoplasms) during routine ultrasonographic surveillance (Fig. 1) [10,24]. In utero diagnosis of SCT warrants vigilant fetal monitoring because of the higher risks of polyhydroamniosis, fetal hydrops, or placentomegaly, which may lead to premature delivery or fetal demise. The diagnosis of SCT prior to a gestational age (GA) of 30 weeks is especially hazardous [4]. Fetal dystocia and rupture of tumors during labor are also concerns of obstetricians. Since SCT is a hypervascular tumor, tumor rupture and massive hemorrhage are possible during labor. Cesarean section may reduce the risks accompanying labor and delivery. However, babies with a small SCT (<5 cm) can be safely delivered vaginally [2]. While a study from the 1980’s reported a high rate of cesarean section (75%), a recent study indicated that 43 out of 44 neonates with SCT were successfully vaginally delivered [11,34].
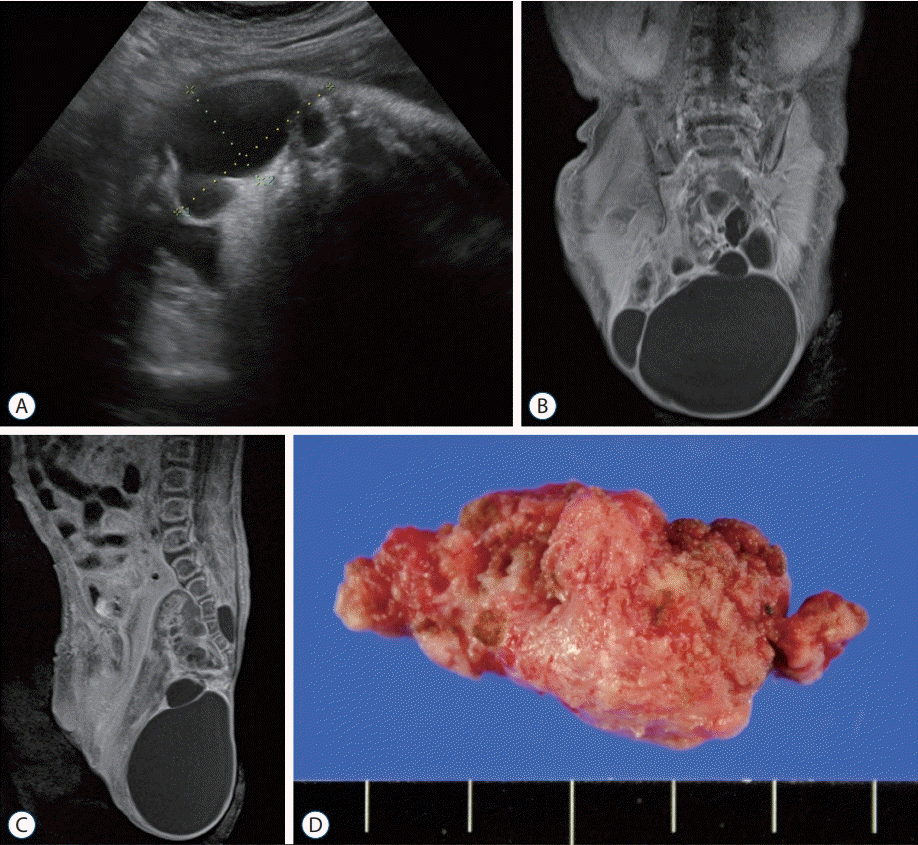 | Fig. 1.Sacrococcygeal teratoma of a female newborn. A : In utero ultrasonography taken at gestational age of 35+3 week reveals a multi-cystic mass protruding outward from the sacrococcygeal area; (B) coronal and (C) sagittal T1-weighted magnetic resonance images show a huge multi-cystic heterogeneous mass located in both intrapelvic and extracorporeal spaces; (D) a gross photograph of the tumor resected at 6th postnatal day.
|
SCTs are usually diagnosed immediately after birth by the presence of a protruding mass. Direct inspection and physical palpation can lead to the appropriate diagnosis [4]. There are anomalies of spinal dysraphism that present with a protruding mass in the sacrococcygeal area, such as cystic limited dorsal myeloschisis, terminal myelocele, and terminal myelocystocele. For differential diagnosis, it is important that SCTs are located more caudally between the anus and coccyx, often displacing the anus anteriorly [47]. Anomalous masses of spinal dysraphism are usually characterized by a higher location at sacrum and coccyx, skin stigmata (hair or hemangioma) over the mass, or accompanying neurological deficits such as lower extremity weakness and bladder dysfunction.
Some SCTs lie entirely within the pelvis, and diagnosis can be delayed for months. Most neonates with SCT are asymptomatic, but large tumors can cause constipation or urinary retention. Abdominopelvic magnetic resonance imaging (MRI) is indicated for all patients to assess the extent of the tumor. Associated malformations such as hip dislocation, spinal dysraphism, and cardiac anomalies should be identified [17]. If lower limb weakness or hypotonia is observed, intradural spinal extension of the tumor or associated spinal dysraphism are considered.
Neonates with a large SCT are at risk of developing consumptive coagulopathy, thrombocytopenia, disseminated intravascular coagulation, and profuse hemorrhage [26]. Therefore, platelet count and coagulation profile should be checked. Serum alpha fetoprotein (AFP) and human β-chorionic gonadotropin (β-hCG) levels aid is the diagnosis of GCT and its subtypes before surgery. Especially, YST can produce high levels of AFP. However, interpretation of the serum AFP value needs caution because in neonates and infants, the levels of serum AFP are normally elevated due to hepatic production [3].
SCT has been classified according to their anatomical location. The Altman classification system divides SCT into four types (Fig. 2) [1]. Type I : tumors predominantly external (45%); type II : tumors presenting externally but with a significant intrapelvic portion (35%); type III : tumors predominantly intrapelvic (10%); type IV : presacral tumors without an external component (10%). Large type II–IV SCT can exert mass effects on intrapelvic organs and present with severe problems such as constipation, fecal incontinence, neurogenic bladder, and urinary incontinence [30]. Large type III SCT often requires extensive abdomino-sacral resection and carries high risks of a poor functional outcome [24].
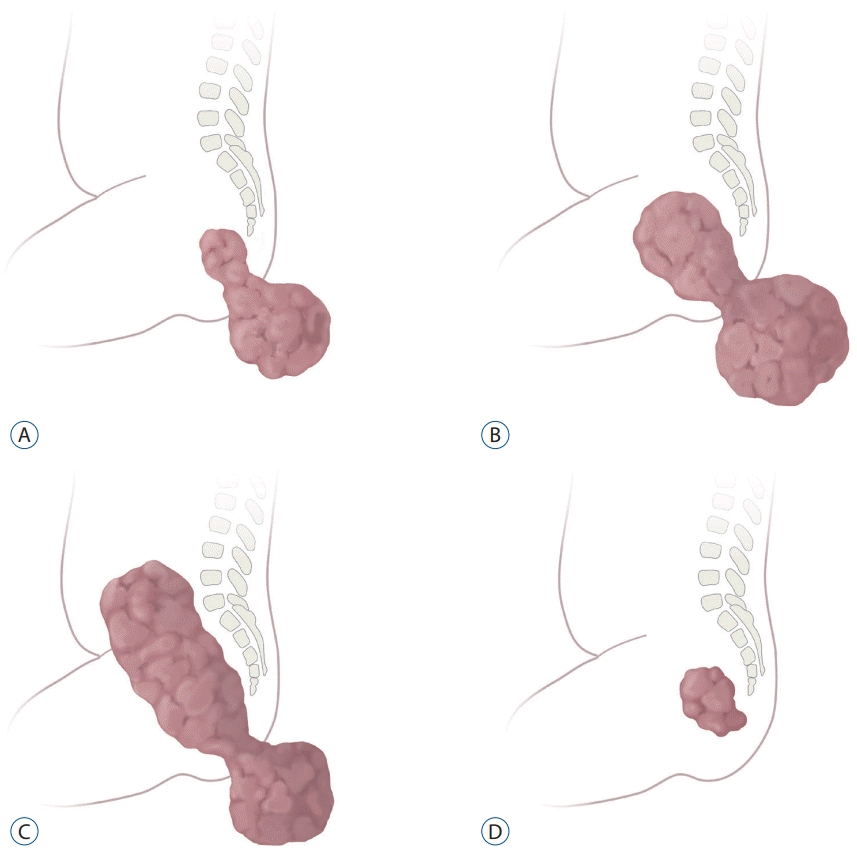 | Fig. 2.Altman classification of sacrococcygeal teratoma according to the anatomical configuration. A : Type I : predominantly external with small internal components. B : Type II : dumbbell-shaped tumor with similar internal and external components. C : Type III : predominantly internal and smaller external components. D : Type IV : exclusively internal (intrapelvic) tumor.
|
Go to :
TREATMENT
Complete surgical resection is the standard for SCT. Delayed treatment may lead to tumor rupture and hemorrhage. SCT has a tendency toward malignant degeneration, which makes early surgery mandatory. In a study, 91% of patients diagnosed at birth had mature/immature teratomas, whereas 27% of infants diagnosed later had YST [35]. The majority of patients (Altman type I and II) can be operated on via the posterior (sacral) approach only. Large type III tumors with both external and internal components may require combined transabdominal and posterior approaches [4]. Purely intrapelvic tumors (type IV) may be approached via laparoscopy [4].
The surgical outcome and prognosis of SCT are favorable. The recurrence rate after surgery has been estimated as 10–15% [23,29,45]. Incomplete resection and immature/malignant histology are the most important risk factors for recurrence [23,45]. Preoperatively, older age at diagnosis (>2 months) and predominantly solid components within the mass are suggestive of malignant histology and a poor prognosis [4,9,47]. To prevent incomplete resection, removing the coccyx bone and preventing tumor tissue spillage during surgery have been emphasized [4,45,47]. Although immature teratoma histology is a risk factor for recurrence, postoperative chemotherapy is not recommended because it is ineffective in preventing recurrence [23]. Recurrent SCT tends to have a malignant histology. One study showed that in eight patients with recurrent SCT, initial mature teratoma recurred as immature teratoma (one patient) and YST (four patients) [45]. The majority of recurrences are diagnosed 6–36 months after the initial surgery [45,46]. Serum AFP is useful in monitoring for recurrences. In a systemic review, serum AFP levels were elevated in 75% of patients with recurrent SCT [43]. Therefore, careful monitoring with digital rectal exam and serum AFP check at 3-month intervals for 3–4 years is generally recommended [4,29]. MRI or computed tomography should be performed if suspicious clinical finding or elevated AFP level is noted [4]. The German study protocols included routine imaging surveillance at 3-month intervals for the first year and at 6-month intervals in the second year [38]. However, long-term surveillance is warranted because patients with a very late recurrence after 5–15 years have been reported [29].
Patients with recurrent SCT can be salvaged by surgery and chemotherapy [46]. Malignant recurrence requires intensive management, and multimodality treatment can establish second and third remissions in many patients [38]. There is no standard chemotherapeutic regimen for malignant recurrent SCT, but it has been emphasized that platinum compound should be included to achieve stable remission [38]. JEB (carboplatin, etoposide, bleomycin) and ICE (ifosfamide, carboplatin and etoposide) regimens were applied to recurrent malignant YST in the UK study [23]. The 5 year overall survival of recurrent, malignant SCT (excluding teratoma recurrence) was 42% [38]. Functional outcome, however, is a concern for survivors. A study reported anorectal sequelae in 29% and urological dysfunction in 33% of survivors [30]. Chronic constipation and fecal incontinence are common anorectal problems, and neurogenic bladder, vesicoureteral reflux, and urinary incontinence constitute common urologic sequelae. A questionnaire survey from the Netherland indicated that long-term survivors of SCT complained of impaired bowel function (46%), urinary incontinence (31%) and unacceptable scarring (40%) [7]. Extensive pelvic surgery in early life also tends to lead to sexual dysfunction in adulthood [17]. Therefore, long-term surveillance and evaluation of functional status are important for the survivor programs for SCT.
In addition to classic surgical resection for SCT, a number of innovative interventions have been developed to rescue complicated fetuses with a large SCT or to decrease surgical bleeding in neonates. Fetal SCT, especially when diagnosed before GA 30 weeks, has a far worse prognosis than neonatal SCT. If the tumor has a large cystic portion, cyst aspiration in utero can prevent preterm labor or reduce fetal dystocia during delivery [12]. Rapid growth of hypervascular, solid SCT causes high output cardiac failure and fetal hydrops, leading to fetal demise. Open in utero tumor debulking is a challenging, but viable option for such cases. Researchers in the Children’s Hospital of Philadelphia reported the early outcome of four patients who received open fetal surgery for SCT [12]. All patients experienced preterm birth after surgery, but three of the four patients have survived without the disease except for one patient who died early. Interventional techniques to ablate feeding arteries to SCT are also under investigation to reverse fetal cardiac failure. Researchers in Toronto tried fetoscopic laser ablation, radiofrequency ablation, and intravascular coiling in five fetuses with SCT [44]. Although they used minimally invasive techniques, only two patients survived after the procedures. Experience from Brazil and France showed similar results of percutaneous interventions for fetal SCT : two out of five patients survived [37]. Therefore, fetal treatments of SCT, open or interventional, are still investigational. However, considering the poor prognosis of fetuses developing cardiac failure and hydrops in utero, these early intervention techniques deserve to be scrutinized and developed.
Interventions can be applied to postnatal patients with large SCTs to reduce the chance of spontaneous hemorrhage, stabilize the cardiopulmonary status, and decrease surgical bleeding. In the 1990s, staged operations, i.e., devascularization surgery followed by tumor resection were tried for patients with huge SCTs [36]. In 2006, Cowles et al. first reported successful embolization of feeding vessels in a newborn of GA 36 weeks with a large hypervascular SCT, followed by similar attempts by others [5,18]. Preoperative embolization seems to be a promising and effective treatment, but the technique needs great expertise and skill. Currently, interventional approaches are not routinely practiced, and only selective centers with such resources can utilize them successfully for SCT [9].
Go to :
 XML Download
XML Download