

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Primary spinal cord tumors (PSCTs) are one of the rarest categories of tumors, accounting for 4–8% of all central nervous system (CNS) tumors [13,17,26]. The incidence of pediatric PSCT (0.26 per 100000 person-years) is even lower than of the incidence among adults (0.74 per 100000 person-years) [28]. In addition to the rare incidence, the pathological presentation of pediatric PSCT varies widely. Because of these obstacles, there exists only literature comprised of a small series of patients [5,8,32] or a larger series of patients in multicenter studies of specific tumor types [15,24].
In the present study, we report a series of 126 pediatric patients with PSCTs who underwent surgery in a single institution. A descriptive analysis regarding symptomatology, tumor level, anatomical compartment, pathological diagnosis, and surgical outcomes was performed. In addition, we conducted detailed survival analysis according to particular anatomical compartments.
MATERIALS AND METHODS
This study was approved by the Institutional Review Board of Seoul National University Hospital (H-1910-168-1074). Between October 1985 and December 2017, a total of 183 consecutive pediatric patients with spinal cord tumors were surgically treated, and their charts were retrospectively reviewed. The clinical presentation, radiographic imaging characteristics, surgical outcomes and pathological results were evaluated. Follow-up was conducted for up to 10 years. Patients with metastatic lesions in the spine were excluded from this study. Patients with multiple craniospinal tumors, usually associated with genetic syndromes such as neurofibromatosis (NF) or von Hippel-Lindau syndrome, were also excluded. Among patients with multiple lesions, most patients (26/27) had NF (NF-1 in nine patients, NF-2 in 14 patients, and undetermined type in three patients). Nonetheless, patients who had solitary spinal lesions in neuraxis were included in the study despite having genetic syndromes (n=5). Following medical chart and radiographic image review, 57 patients were excluded (metastasis in 25 patients, multiple craniospinal tumors in 27 patients, and incomplete data in five patients). Finally, a total of 126 patients were evaluated for the present study.
Magnetic resonance imaging (MRI) was adopted at our institution in September 1987. Before the adoption of MRI, PSCTs were diagnosed using computed tomography-myelography. The number of involved segments, involved spinal level, and anatomical compartment of the tumor were analyzed. The level of tumor involvement was classified into cervical, cervico-thoracic, thoracic, thoraco-lumbar, lumbar, lumbosacral, and sacral. Tumors involving almost the whole spinal cord were particularly classified as holocord tumors. The anatomical compartment was categorized into intramedullary (IM), intradural extramedullary (IDEM), and extradural (ED) compartments. Some ED tumors with extension to the paravertebral space were also categorized as ED tumors. Tumors extending beyond one anatomical compartment were described by including the whole area of the tumors (IDEM/ED, for example).
The extent of tumor removal was categorized into gross total removal (GTR; >95% of tumor removal), subtotal removal (STR; >80% of tumor removal), partial removal (PR; <80% of tumor removal), and biopsy [11,33]. The mode of bony removal was categorized into laminoplasty, laminectomy, (partial) hemilaminectomy, and corpectomy.
All tumor specimens were inspected by a neuropathologist. The classification of tumors was based on the 2016 World Health Organization (WHO) classification of tumors of the CNS [18]. Tumor biological behaviour codes /0 and /1 were classified as nonmalignant tumors, whereas tumors coded /3 were classified as malignant tumors. ID tumors were classified by a grading system. The old pathologic specimens were reviewed, and a molecular study was performed if possible.
The patient’s neurological status was assessed preoperatively and postoperatively at three months using the modified McCormick scale. The modified McCormick scale is as follows : grade I, neurologically intact, ambulates normally, may have minimal dysesthesia; grade II, mild motor or sensory deficit, patient maintains functional independence; grade III, moderate deficit, limitation of function, independent of external aid; grade IV, severe motor or sensory deficit, limitation of function with a dependent patient; and grade V, paraplegia or quadriplegia, even if there is flickering movement [20]. Progression-free survival (PFS), defined as the absence of any clinical or radiographic sign of recurrence of the tumors, and overall survival (OS) were estimated. OS and PFS were evaluated for total PSCT patients as well as for the IM and ED groups. Tumors spanning more than one anatomical compartment (such as IM/IDEM and IDEM/ED) were not analyzed separately for survival statistics.
To assess survival data for patients who were lost to follow-up, we requested and collected death registry data from the National Statistical Office.
Statistical analysis
Statistical analysis was performed using IBM SPSS ver. 22.0 software for Windows (IBM Corp., Armonk, NY, USA). The distributions of the variables are described as the mean value and standard deviation. Independent t-tests and paired t-tests were used to compare parametric variables before and after the operation. The degree of functional improvement after surgery was evaluated utilizing a linear mixed model. The Mann-Whitney test was utilized to analyze nonparametric independent variables. PFS and OS were assessed with the Kaplan-Meier technique. Survival analysis was conducted according to extent of removal as well as pathology. Survival analysis according to pathology for total PSCT and ED tumors was performed in terms of malignancy (malignant vs. benign). For IM tumors, on the other hand, the analysis according to pathology was performed in terms of grade (high grade vs. low grade). Statistical significance was set at p<0.05.
RESULTS
Patient demographics
There were 126 pediatric patients enrolled in this study (Table 1). The number of male patients and female patients was 56 and 70, respectively. The mean age of the patients was 6.4±5.04 years. The mean symptom duration was 9.3±21.61 months. The symptom duration was significantly shorter in patients with malignant tumors than in patients with benign tumors (5.5±17.42 months vs. 13.6±25.27 months, p=0.038). In terms of sex and age, the symptom duration did not show significant differences. The mean follow-up period was 69.5± 46.30 months. The presenting symptoms were categorized into motor deficit, sensory disturbance, urinary disturbance, spinal deformity, soft tissue mass, radiographic finding, and miscellaneous. The most common presenting symptom was motor deficit (n=101, 80.2%). Among the motor symptoms, weakness of extremities (n=69) was the most common, followed by gait disturbance (n=28). Sensory disturbance was the second most common symptom type (n=40, 31.7%), and pain was the most common sensory disturbance (n=33). Urinary disturbance was present in nine patients (7.1%). Ten patients (7.9%) were diagnosed with PSCTs during detailed imaging studies for evaluation of spinal deformity. An incidental mass on radiographic studies for irrelevant symptoms was found in 16 patients (12.7%). Miscellaneous symptoms were present in seven patients (5.6%).
In 43 patients with IM tumors (male : female, 16 : 27), the mean age of patients was 6.9±5.18 years. The mean period of symptom duration until diagnosis was significantly longer than that in patients with ED tumors (17.5±32.50 months vs. 2.2±4.12 months, p=0.004). The mean follow-up period was 63.1±48.32 months. Motor deficits were more common in patients with IM tumors than in patients with ED tumors (97.7% vs. 59.6%, p<0.001). Additionally, there were significantly more patients presenting with spinal deformity with IM tumors than with ED tumors (16.3% vs. 0%, p=0.003).
In 57 patients with ED tumors (male : female, 26 : 31), the mean age of patients was 5.7±5.10 years. The mean follow-up period was 72.4±47.01 months. The proportion of patients diagnosed with incidental masses on radiographs was significantly higher among patients with ED tumors than among patients with IM tumors (26.3% vs. 0%, p<0.001).
Radiographic characteristics
The most common anatomical compartment was ED (n=57, 45.2%), followed by IM (n=43, 34.1%) and IDEM (n=16, 12.7%, Table 2). Among ED tumors, 12 cases (21.1%) were located exclusively epidurally within the spinal canal. The remainder (n=45, 78.9%) involved not only the epidural space but also the paravertebral space. Tumors spanning more than one anatomical compartment were as follows : IM/IDEM (n=2, 1.6%) and IDEM/ED (n=8, 6.3%).
The mean number of involved segments was 5.4±4.61. The most commonly involved level was the thoracic spine (n=40, 31.7%). Holocord involvement of the tumor was found in nine patients (7.1%). In IM tumors, the involved segments were significantly longer than those of ED tumors (8.5±5.69 vs. 3.8± 3.03, p<0.001). Additionally, holocord involvement (18.6%) was significantly more predominant in IM tumors than in ED tumors (8/43 vs. 1/57, p=0.010). The most commonly involved levels were the cervicothoracic spine (n=12, 27.9%) and thoracic spine (n=30, 52.6%) in IM tumors and ED tumors, respectively.
Surgical treatment
Regarding the extent of tumor removal (Table 3), GTR was achieved in 63 patients (50.0%). STR and PR were achieved in 41 (32.5%) and 14 (11.1%) patients, respectively. In eight patients (6.3%), open biopsy was conducted. In patients with IM tumors, GTR and STR were each achieved in 16 patients (37.2%). In patients with ED tumors, GTR was achieved in 33 patients (57.9%), and STR was achieved in 17 patients (29.8%).
Regarding operative methods (Table 3), the most commonly performed procedures were laminoplasty (n=70, 55.6%) and laminectomy (n=36, 28.6%). Both in patients with IM tumors and ED tumors, the majority of patients underwent laminoplasty (65.1% in IM tumors and 42.1% in ED tumors).
Tumor pathology
Among the 126 patients, 69 patients had malignant tumors (54.8%, Table 4). The proportion of malignancy was highest in ED tumors (40/57, 70.2%); on the other hand, malignant tumors were absent in IDEM tumors (0/16). Among IM tumors (n=43), high-grade tumors were confirmed in 11 patients (25.6%). The most common pathologies were schwannomas (n=14) and neuroblastomas (n=14). Regarding pathology, schwannomas were the most common benign tumors, whereas neuroblastomas were the most common malignant tumors. In terms of anatomical compartment, diffuse astrocytomas (n=7), neuroblastomas (n=14), and schwannomas (n=8) were the most common pathologies among IM, ED, and IDEM tumors, respectively.
Regarding the WHO classification of CNS tumors, 95 out of 126 tumors (75.4%) were classified as CNS tumors. Among them, the most common type was tumors of the paraspinal nerves (n=23, 18.3%), including schwannomas (n=14), neurofibromas (n=7), and malignant peripheral nerve sheath tumors (MPNST, n=2). The second most common type of tumor was mesenchymal, nonmeningothelial tumors, which were confirmed in 19 patients (15.1%). Among them, Ewing sarcomas (EWS) were the most common (n=10). Regarding six specimens of EWS that were able to be genetically studied, all specimens revealed EWS translocation. CNS embryonal tumors were identified in five patients (4.0%) : atypical teratoid/rhabdoid tumors (AT/RT) in three patients, all of which revealed INI1 alteration, and CNS embryonal tumors with rhabdoid features in two patients.
Thirty-one out of 126 tumors (24.6%) could not be classified into CNS tumors. These tumors were peripheral neuroblastic tumors, including neuroblastomas (n=14), ganglioneuromas (n=12), and ganglioneuroblastomas (n=5).
Among IM tumors (n=43), the most common tumors were diffuse astrocytic and oligodendroglial tumors (n=17, 39.5%), including diffuse astrocytomas (n=7), anaplastic astrocytomas (n=4), glioblastomas (n=3), oligodendrogliomas (n=2), and anaplastic oligodendroglioma (n=1). There was no case of diffuse midline glioma characterized by K27M mutations in the histone H3 gene H3F3A. Pilocytic astrocytomas, categorized as other astrocytic tumors, were found in six patients (14.0%). In nine patients (20.9%), neuronal and mixed neuronal-glial tumors were found. There were six cases (14.0%) of mesenchymal and nonmeningothelial tumors. Ependymal tumors were confirmed in three patients (7.0%).
Among ED tumors (n=57), the most common type was peripheral neuroblastic tumors (n=31, 54.4%) : neuroblastomas (n=14), ganglioneuromas (n=12), and ganglioneuroblastoas (n=5). Mesenchymal, nonmeningothelial tumors were the second most common type of ED tumors (n=10, 17.5%), consisting of EWS (n=9) and hemangioma (n=1). There were six cases (10.5%) of tumors of the paraspinal nerves, including schwannomas (n=3), neurofibroma (n=1), and MPNSTs (n=2).
Clinical outcomes
The preoperative modified McCormick scale was 2.8±1.34, which improved to 2.2±1.42 postoperatively (p<0.001). The postoperative modified McCormick scale improved in 55 patients (43.7%) compared to the preoperative condition. On the other hand, the modified McCormick scale worsened postoperatively in 13 patients (10.3%). In 56 patients (44.4%), the perioperative modified McCormick scale remained unchanged. The modified McCormick scale could not be assessed in two patients.
The postoperative modified McCormick scale was also significantly improved in both the group of patients with IM tumors (3.1±1.21 preoperatively vs. 2.7±1.31 postoperatively, p=0.007) and the group of patients with ED tumors (2.5±1.49 preoperatively vs. 1.8±1.36 postoperatively, p<0.001). Preoperative functional status in patients with IM tumors, assessed utilizing the modified McCormick scale, showed a trend towards being worse than that in patients with ED tumors (3.1±1.21 vs. 2.5±1.49), without statistical significance (p=0.074). The postoperative modified McCormick scale of patients with IM tumors was significantly worse than that of patients with ED tumors (2.7±1.31 vs. 1.8±1.36, p=0.002). The degree of improvement in the modified McCormick scale after surgery showed no significant difference (p=0.156) between patients with IM tumors and ED tumors (Fig. 1).
Functional outcomes were evaluated in terms of the extent of tumor removal. In patients with IM tumors, the modified McCormick scale improved in both patients with GTR (3.3±1.23 preoperatively, 2.7±1.27 postoperatively) and non-GTR (2.9±1.27 preoperatively, 2.6±1.39 postoperatively) after surgery. The degree of improvement of the modified McCormick scale showed an insignificant difference regarding the extent of tumor removal (p=0.447).
Similar to patients with IM tumors, the modified McCormick scale also improved in patients with ED tumors who achieved GTR (2.3±1.40 preoperatively, 1.5±1.19 postoperatively) and non-GTR (2.8±1.57 preoperatively, 2.1±1.51 postoperatively) after surgery. The degree of improvement of the modified McCormick scale did not show a significant difference regarding the extent of tumor removal (p=0.812).
Twenty-two patients (17.5%) died of the disease. The mean time until death was 15.8±15.85 months. The most common pathology of patients who died from the disease was malignant tumors (21/22, 95.5%) : EWS (n=7), anaplastic astrocytoma (n=3), CNS embryonal tumor with rhabdoid features (n=2), AT/RT (n=2), glioblastoma (n=2), oligodendroglioma (n=1), lymphoma (n=1), neuroblastoma (n=1), peripheral neuroepithelioma (n=1), and MPNST (n=1). There was one patient with a benign tumor who died from the disease. The patient underwent PR (total resection of intraspinal portion) of an ED neurofibroma with substantial extraspinal involvement. After 5 years, he was diagnosed with malignant transformation of neurofibroma (MPNST) and died from the disease (survival period of 66 months). Patients who died of the disease had a significantly shorter duration of symptom presentation until diagnosis (1.7±2.62 months vs. 11.1±23.62 months, p<0.001), worse preoperative modified McCormick scale (3.6±1.50 vs. 2.6±1.24, p=0.001), and worse postoperative modified McCormick scale (3.5±1.50 vs. 1.9±1.23, p<0.001) than those who survived. The age, sex, and number of involved segments did not show statistical significance between patients who died and those who survived.
Postoperatively, 36 patients (28.6%) suffered from disease progression. The mean period of progression was 22.6±30.81 months. Patients who showed disease progression had a significantly shorter symptom duration than patients without progression (2.5±4.09 months vs. 12.3±25.24 months, p=0.001). There was no significant difference between patients with and without disease progression in terms of age, sex, or number of involved segments. Among patients with disease progression, 15 patients (41.7%) underwent revision surgery. After revision surgery, three patients received both chemotherapy and radiotherapy, two patients received chemotherapy, and one patient underwent proton therapy for recurrent myxopapillary ependymoma. Five patients (13.9%) were treated with chemotherapy, radiotherapy, or both without revision surgery. Sixteen patients (44.4%) did not receive any treatment for disease progression. Seven patients could not receive additional treatment because of rapid disease progression or poor general condition. Four patients rejected receiving any treatment and received hospice care. Two patients were undergoing imaging follow-up for slow progression. Three patients were lost to follow-up.
Of patients with IM tumors, eight patients (18.6%) died from the disease. The median time until death was 11 months (20 days to 26 months). The majority of patients who died, except one patient, had high-grade tumors : anaplastic astrocytomas (n=3), glioblastomas (n=2), EWS (n=1), malignant lymphoma (n=1), and oligodendroglioma (grade II, n=1). Sixteen patients (37.2%) suffered from disease progression. The median period of progression was 7.5 months (20 days to 114 months). Most of the disease progression occurred within 2 years, except in two patients. One patient with diffuse astrocytoma showed disease progression at 72 months. Because the patient did not undergo additional operation or detailed radiographic studies, we could not determine whether malignant transformation occurred. Another patient with myxopapillary ependymoma showed radiographic recurrence at 114 months. She underwent revision surgery with subtotal resection of the tumor and continued regular follow-up. Among patients with disease progression, eight patients (50%) received additional treatment, including surgery, chemotherapy, or radiotherapy; five patients (31.3%) received only supportive care; one patient (6.3%) is on regular imaging follow-up; and two patients (12.5%) were lost to follow-up.
Of the patients with ED tumors, 10 patients (17.5%) died from the disease. The mean time for death was 17.1±13.70 months. The pathological results of patients who died all indicated malignant tumors. Most deaths occurred within 2 years, except for in one patient (50 months). Ten patients (17.5%) suffered from disease progression. The median period of progression was 12 months (13 days to 40 months). Among patients with disease progression, six patients (60%) were treated with surgery and/or chemotherapy. Three patients (30%) did not receive any treatment because of poor general condition or refusal of treatment, and one patient was lost to follow-up.
OS and PFS
The 5- and 10-year OS rates were 82% and 81%, respectively (Fig. 2, Supplementary Table 1). The 5- and 10-year PFS rates were 73% and 66%, respectively. Patients with malignant tumors showed significantly worse survival statistics than patients with benign tumors (p<0.001). The PFS also showed significant differences in terms of pathology (p=0.031). In terms of the extent of tumor removal, the patients with GTR showed a significantly better survival curve than those without GTR (p<0.001). PFS was also better in patients with GTR than in patients without GTR (p=0.003).
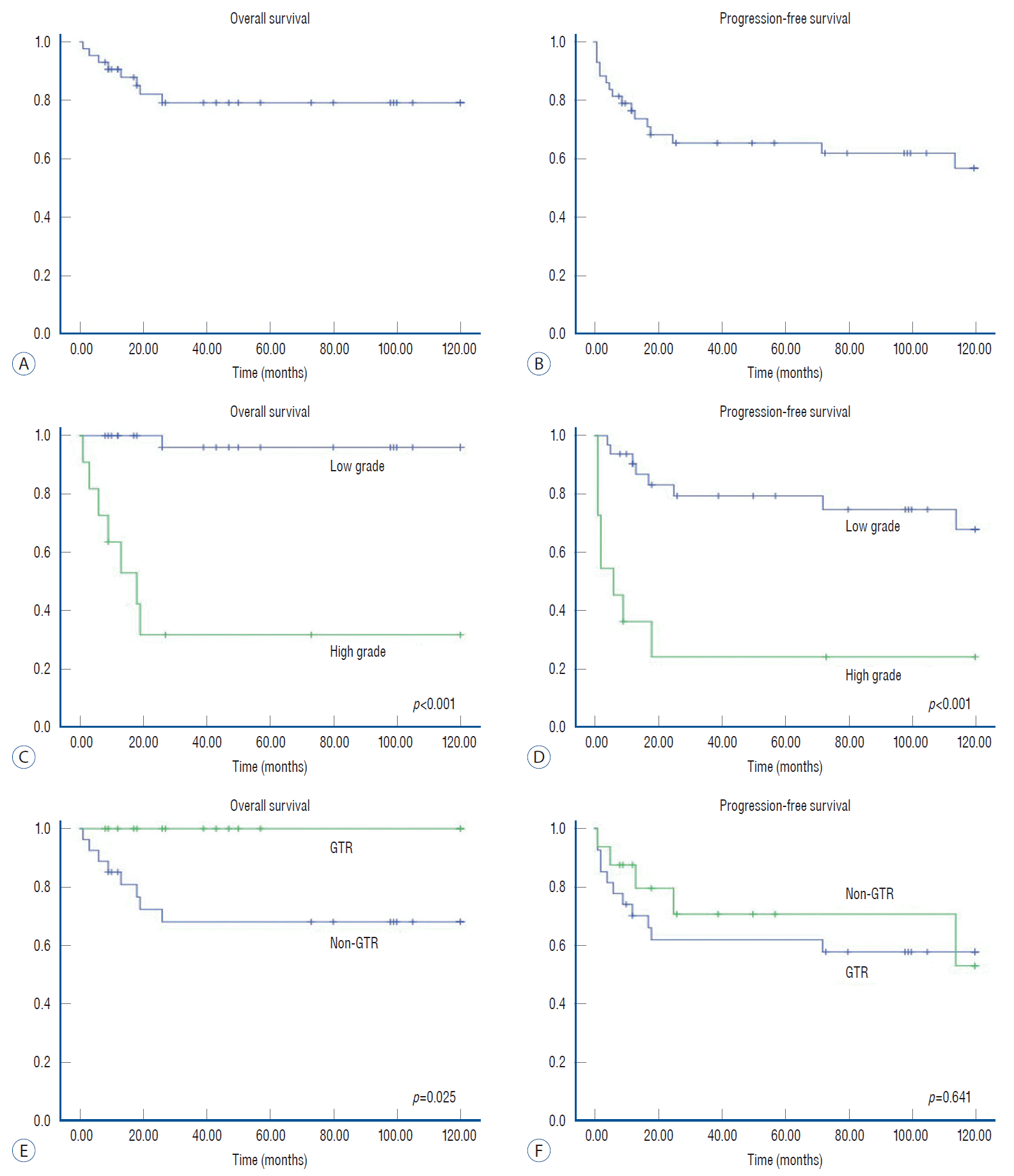
Among patients with IM tumors, the 5- and 10-year OS rates were 79% and 79%, respectively (Fig. 3). The 5- and 10-year PFS rates were 65% and 57%, respectively. Tumor grade showed significant differences in regard to survival (p<0.001) and progression (p<0.001). In terms of the extent of resection, all patients who achieved GTR of IM tumors survived. However, the OS rates in patients without GTR were 68% at 5 and 10 years (p=0.025). The PFS in patients with IM tumors, however, did not show a significant difference regarding the extent of resection (p=0.641).
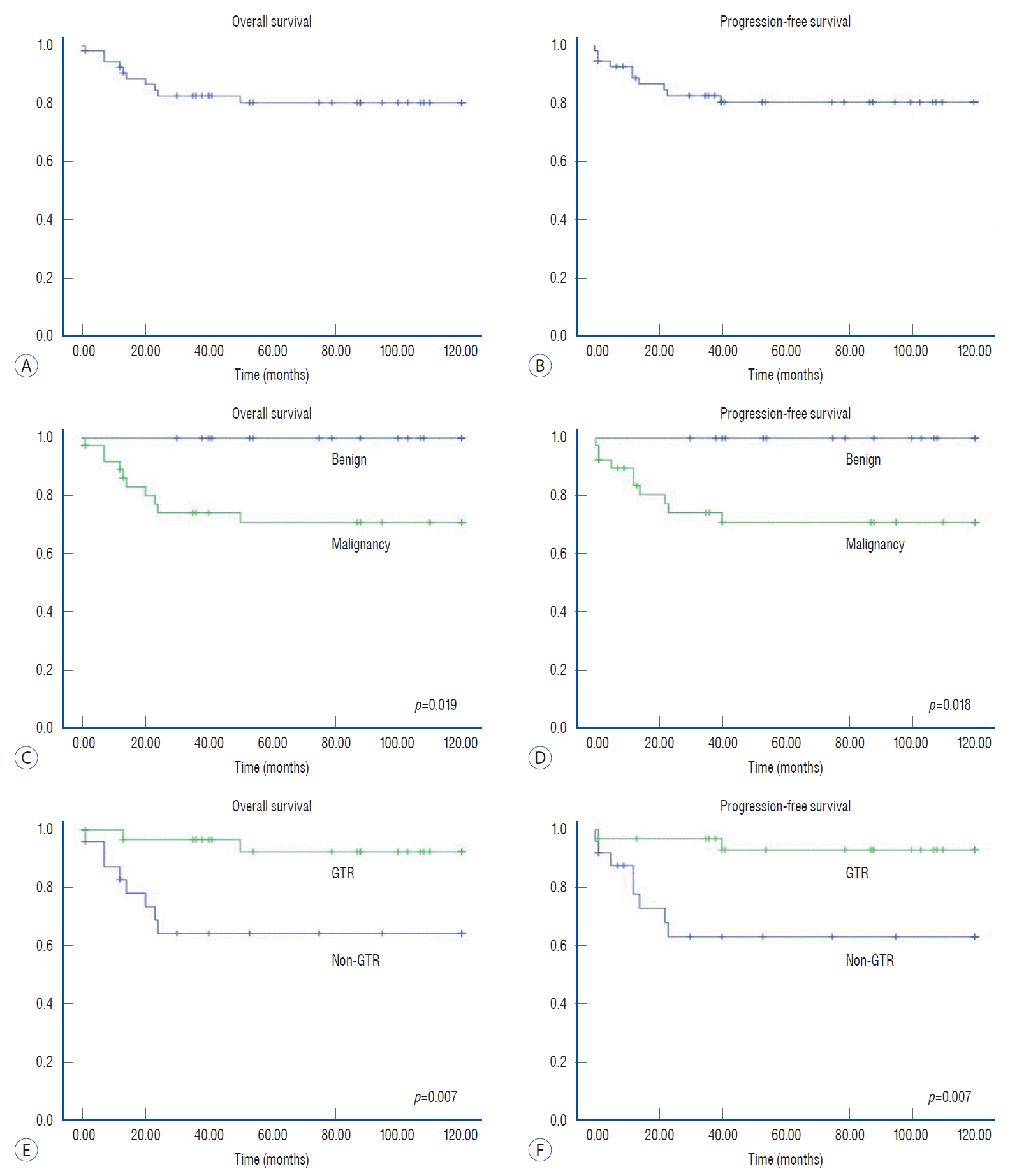
Among patients with ED tumors, the 5- and 10-year OS rates were 80% and 80%, respectively (Fig. 4). The 5- and 10-year PFS rates were 81% and 81%, respectively. Tumor pathology also showed significant differences in regard to survival (p=0.019) and progression (p=0.018). Unlike patients with IM tumors, the extent of removal had a significant effect on OS (p=0.007) and PFS (p=0.007) in patients with ED tumors.
DISCUSSION
In the present study, we reviewed a 32-year experience of surgical management of pediatric PSCT in a single institution. Although several studies comprising a large number of pediatric patients with IM PSCTs exist, studies regarding tumors involving entire spinal columns are quite rare [9,32]. To the best of our knowledge, the present study is the largest series comprising all primary neoplasms of the spinal axis with an evaluation of detailed survival analysis.
The mean duration of symptoms was 9.3 months in the present study. This is similar to most previous studies ranging from 6 to 12 months [8,11,12,15,16,29]. As Houten and Weiner [15] described, the symptom duration was significantly shorter in patients with malignant tumors in our study. It is noteworthy that the symptom duration was much shorter in patients with ED tumors (2.2 months) and longer in patients with IM tumors (17.5 months) than in the total study population. These differences might result from the high proportion of malignancy in ED tumors (70.2%) and the predominance of lower grade tumors in IM tumors (74.4%).
The majority of children diagnosed with PSCT presented with some type of symptom. Among the symptoms, motor weakness (80.2%), including gait disturbance, was most common. This is in contrast to some previous studies that described that the most common presenting symptomatology was neck or back pain [12,15,29,30]. However, others described motor weakness as the most common symptom [7,8,11,16,21]. Despite the discrepancy in dominant symptoms, the sensory symptoms in pediatric patients with spinal cord tumors are not as predominant as those in adult patients [8,23]. This may be because pediatric patients often cannot be aware or complain of sensory change, while adolescents or adults can [8]. Among the patients presenting with spinal deformity, most of them (7/10) had IM tumors. The prevalence of spinal deformity in patients with IM tumors (7/43, 16.3%) was similar to that in previous literature [1,32,33]. As described earlier, spinal deformity was more evident in patients with IM tumors than in patients with tumors in other compartments because of the inequality between signalling to both sides of the spine and paraspinal muscles [3]. There have been increasing issues related to postoperative progressive spinal deformity following laminectomy and irradiation [27,32,33]. Post-laminectomy kyphoscoliosis is beyond the scope of the present study; however, further studies are necessary. In ED tumors, the proportion of incidental masses on radiographic examination was significantly higher than that in IM tumors (26.3% vs. 0%, p<0.001). The high incidence of peripheral neuroblastic tumors among ED tumors characterized by large mediastinal or retroperitoneal masses could explain the incidental findings.
In contrast to the adult type, pediatric PSCTs tend to extend to multiple levels [22,30]. In the present study, approximately half (56.3%) of patients had tumors involving more than three levels, and nine patients (7.1%) revealed holocord tumors, which was similar to the previous literature [30]. Regarding the anatomical compartment, ED tumors were most common (45.2%), followed by IM tumors (34.1%) and IDEM tumors (12.7%) in the present study. The anatomical distribution of PSCTs was quite similar to those described in previous studies. Spacca et al. [30] reported in their study consisting of 134 patients that the anatomical compartment was ED in 39.5% and IM in 34.3% of patients. Wetjen and Raffel [31] reported ED tumors accounted for 34.5% and IM tumors accounted for 29.7% by collecting and assessing 10 studies. Although some studies reported that the IM compartment was most common, it seems obvious that, unlike adult counterparts, pediatric IDEM tumors constitute a small portion of PSCTs [3,12].
Regarding pathologic presentation, malignant tumors comprise half of all tumors. However, the proportion of malignancy significantly differs in terms of anatomical compartment. Although malignant tumors account for approximately half of IM tumors (tumors’ biologic behaviour code /3 in WHO classification of CNS tumors), approximately 70% of ED tumors are malignant. There was no malignant tumor in the IDEM location. These findings imply that PSCTs have distinct behavioural characteristics according to anatomical compartment and should be assessed separately. Similar to previous studies, the most common types of tumors were nerve sheath tumors (n=23), low-grade gliomas (n=20), and neuroblastomas (n=14). Astrocytic tumors such as pilocytic astrocytomas, diffuse astrocytomas, anaplastic astrocytomas, and glioblastomas were dominant among IM tumors (46.5%) [7,11,30].
Notably, the pathologic composition of pediatric spinal cord tumors is far different from that of adult tumors [19]. First, the majority of spinal cord tumors in adults are metastatic tumors. In the present study, however, approximately one-quarter (36/126) of all tumors were embryonal tumors (CNS or peripheral origin), and there were only 25 patients out of 183 patients on registry (before exclusion) with metastatic lesions (13.7%). Moreover, the majority of cases of metastatic lesions occurred from metastasis from the brain, not from solid organs. Second, meningiomas are encountered very rarely, in contrast to in adults, in whom meningiomas are one of the most common tumors [28]. There was no case of meningioma in the present study after exclusion of one patient with atypical meningioma because of multiple neuraxis tumors associated with NF-2. Furthermore, summing all cases of meningiomas in the literature about pediatric PSCT, there were only three cases among 255 patients (1.2%) [8,12,30,32]. Although the old research by Fortuna et al. [14] stated that meningiomas comprised 4.3% of childhood spinal tumors (sporadic in 21 cases, NF-associated in five cases), it seems much rarer than previously reported. Third, the histologic proportion in IM tumors also differs from that in adult IM tumors. Astrocytomas predominate over ependymomas, especially in younger children; gangliogliomas are more prevalent; and hemangioblatomas are found very rarely in the pediatric population [10,15,25]. Our study regarding IM tumors was also consistent with previous literature. Nonetheless, the proportion of ependymomas (including IM myxopapillary type) in IM tumors was very low (n=3, 7%) in the present study. Spinal cord ependymomas are known to be associated with NF-2 in the pediatric population, and mutations in the NF-2 transcript are frequently found even in sporadic cases [3,6]. Before exclusion, there were eight cases of IM ependymomas, which were the second most common IM tumors, as previously reported [3,11,12,30]. However, five cases of ependymomas were excluded from the study because of multiple lesions associated with NF-2 (n=4) and incomplete data (n=1).
Regarding clinical outcomes, 36 patients (28.6%) suffered disease progression, and 22 patients (17.5%) died from the disease. According to previous literature, progression rates and mortality rates of pediatric PSCTs range from 18.5–37.4% and 8.8–40.0%, respectively [5,8,29,30]. Regarding IM tumors, the 5-year OS rates (82%) and PFS rates (73%) in the present study were quite similar to those of a previous study reported by Constantini et al. [11] (OS rates, 76%; PFS rates, 71%). Consequently, the clinical outcomes of our study were in general agreement with those of previous studies, although literature with detailed survival statistics was hardly found due to the great diversity of spinal cord pathology. In the present study, we conducted survival analysis for the total group and subgroup analysis for IM and ED tumors separately. The major finding in the survival analysis was that patients with benign pathology who achieved GTR of tumors showed significantly better OS rates in the total group and in IM tumors and ED tumors separately.
The benefit of benign pathology on PFS rates was also obvious for total PSCTs, IM tumors, and ED tumors. Similarly, regarding the extent of resection, GTR of tumors showed significantly better PFS rates in the total group and the ED tumor group. For IM tumors, however, the PFS curve demonstrated a different pattern. The beneficial effect of GTR, which was statistically insignificant, completely disappeared after longterm follow-up in the IM tumor group. According to previous literature, the pathological composition is the main determinant for patient survival and tumor progression for pediatric IM tumors [11]. However, the effect on OS and PFS of radical resection of IM tumors seems less clear [11,15,16], although some researchers advocated beneficial effects of total excision of tumors [2,4,8,12]. Constantini et al. [11] reported that long-term PFS did not differ between groups with GTR and STR beyond 3 years postoperatively. These findings were similar to those of the present study. Therefore, for IM tumors, tumor grade seems to be a main determinant of disease progression, whereas the extent of removal had little effect.
It is noteworthy to consider the role of radical excision of tumors in patients with IM tumors. Although the role of GTR was limited in terms of tumor progression, GTR resulted in better OS rates in patients with IM tumors. If the GTR results in profound neurological deterioration after surgery, the attempt to achieve GTR may be questionable. In the present study, however, patients with IM tumors who achieved GTR did not show worse functional outcomes than patients who underwent moderate excision. Consequently, radical excision of IM tumors could be a reasonable option if feasible.
There are some limitations in the present study. First, the main limitation is the heterogeneity of pathology. This heterogeneity is inherent to pediatric PSCTs due to the rarity of this type of tumor, which is the main obstacle to the establishment of a treatment protocol. To overcome this limitation, we categorized and evaluated the tumors according to a particular anatomical compartment. However, we think that future multicenter studies with a larger number of patients are needed. Second, a number of patients were excluded from the study. Representatively, we excluded patients with multiple lesions along the craniospinal axis because the assessment of recurrence or cause of death may be confusing in those patients. We suppose these patients would have worse clinical outcomes than patients with solitary lesions. It would be beneficial to evaluate syndromic patients with multiple lesions separately afterward. Third, there are missing data due to the loss of medical charts or radiographic images before the installation of electronic medical records, picture archiving and communication systems in our institution. The lost data could result in bias in the evaluation. Fourth, because of some old data from before the era of practical use of MRI, in-depth radiographic analysis was not possible. This resulted from the enrolment of an earlier registry to evaluate the long-term outcomes of PSCTs.
CONCLUSION
PSCTs are uncommon in pediatric patients. The most common symptomatology was motor weakness, and the symptom duration was shorter in patients with malignant tumors. Malignancy was found in approximately half of all tumors, the proportion of which is different according to the anatomical compartment. The most common anatomical compartment was ED, followed by IM and IDEM. The most common tumors were schwannomas and neuroblastomas. Both the pathology and extent of resection had a decisive effect on OS. The extent of removal also showed a beneficial effect on PFS in the total group and ED tumors. In IM tumors, however, pathology seems to be a main determinant of PFS, whereas the extent of removal had little effect. Radical excision of IM tumors could be a viable option for survival improvement without an increased risk of worse functional outcomes.


 XML Download
XML Download