

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
‘It is not birth, marriage or death but gastrulation which is truly the most important time of your life’
This statement by the English embryologist Lewis Wolpert serves to emphasise the magnitude and significance of the early embryonic events that constitute the period of gastrulation. These, evolutionarily conserved events, occurring in the third week of development, transform the bilaminar disc into a primitive embryo that not only contains the three germ cell layers of ectodermal, mesoderm and endoderm but has polarity in the craniocaudal, dorso-ventral and left-right dimensions. Until relatively recently it had been assumed that primitive cells were pre-programmed, their destiny in the future individual already defined through the genetic code. Whilst the process of gastrulation remains poorly understood in humans, studies in animal models have begun to reveal a picture of elegant complexity at the cellular level through which identical cells are transformed in response to both physical and chemical factors. In this scenario cells do not “know” the final form of the embryo, rather their genetic code contains a set of instructions, a “generative programme” (Wolport) that leads to changes in cell shape and movement. Cellular movements, combined with direct cell-cell interactions and response to signalling factors results in a co-ordinated behaviour of cells within sheets, these sheets of cells expand and fold, transforming the two dimensional symmetry of the bilaminar disc to the complex three dimensional asymmetry of the post gastrulation embryo. Wolpert suggested that there were four properties of cells that underpinned the process of gastrulation namely, multiplication, movement, changes in cell shape and cell-cell signalling [32].
Events immediately prior to gastrulation
Soon after fertilisation the embryo comprises a sphere of identical embryonic cells. The conundrum that has faced developmental biologists is how do such genetically identical cells soon develop positional and morphological identity? Whilst a complete explanation remains elusive interesting advances in molecular biology have led to the emergence of new concepts [23]. Firstly, it is known that the expression of many thousands of genes within a cell results in a certain ‘randomness’ and therefore variation between cells. This intrinsic ‘noise’ in gene expression can be mathematically modelled and is referred to as stochastic gene expression. Secondly, cells do not exist in isolation but in close proximity to one another and the changing physical constraints imposed by contact creates external ‘noise’. As cells of the morula undergo further division and the sphere of cells increases in size an important physical difference separates the constituent cells into those that remain entirely surrounded by like-cells, and those that have a surface exposed to the outside. There is now evidence to suggest that it is the different physical forces exerted on these outer cells caused by this geometric consequence of growth (rather than ‘pre-programming’) that seems important in trigering a series of intracellular events that activates genes within these ‘outer’ cells, transforming them into the trophoectoderm. These trophoectoderm cells create a shelllike layer that will ultimately differentiate into the placental membranes; these cells secrete fluid into the sphere, which as a result becomes ‘hollowed out’ and known as the blastocyst. Those cells within the original sphere, that lacked the surface exposure, and therefore whose genes were not activated along the trophoectoderm path, now constitute a clump of cells on the inner surface of the hollowed sphere known as the inner cell mass.
Again, there will be some cells of the inner cell mass that have a surface that facing the lumen of the sphere and, as a result undergo changes that transform them into a distinct layer, the hypoblast. Peripheral cells of the hypoblast migrate around the inner surface of the trophoblast shell, transforming the cavity into the definitive yolk sac. The remaining, deeper and non-lumen facing cells of the inner cell mass retain an attachment to the hypoblast but form a distinct layer, the epiblast, which will also separate away from the surrounding shell creating a ‘second space’ within primitive embryo, the amniotic cavity.
As a result of these changes, and immediately prior to gastrulation the primitive embryo consists of a bilaminar disc, a layer each of hypoblast and epiblast facing the yolk sack and amniotic cavity respectively. It is the epiblast layer that, through the processes of gastrulation will provide all the cells of the three primitive germ-cell layers; ectoderm, mesoderm and endoderm.
It will be evident from the above description that current evidence suggests that the fate of individual cells prior to gastrulation has not been pre-determined but occurs in response to physical changes in cell shape dictated by the relative position of cells within a larger structure. It is these physical changes, and the transmembrane changes that they cause, which permits the activation of specific genes able to transform the cell for particular function. The importance of direct cell-cell interactions, signalling factors and chemical gradients within the embryo will continue to be important factors in conferring positional identity and the fate of cells during gastrulation.
The events of gastrulation
Prior to gastrulation the bilaminar disc exhibits radial symmetry and lacks any evidence to suggest the future craniocaudal axis. In animal models, cells of the hypoblast appear to be important in initiating a series of changes that will disrupt this radial symmetry and give the embryo cranio-caudal and left-right polarity. Cells from within the hypoblast (again, likely in response to their relative position within the disc) undergo gene activations that allow them to migrate to a point on the edge of the hypoblast where they aggregate, a site designated as the anterior visceral endoderm (AVE). As already mentioned, it is the epiblast that will give rise to all tissues of the future embryo and at this early stage it is important that the epiblast cells are prevented from differentiating too early. This is achieved through inhibitory signalling from the AVE that maintains the epiblast in a pluripotent state, however, with continued growth, some cells of the epiblast will eventually escape this inhibitory effect, allowing genes to be activated that will facilitate the production of signalling proteins that will induce the movement of surrounding epiblast cells toward a central zone, the primitive streak [31]. The arrival and ‘intercalation’ of successive waves of epiblast cells at the primitive streak marks the beginning of transformation from bilaminar to trilaminar disc and results in lengthening of the primitive streak and elongation of the entire embryo. The first part of the primitive streak to form is the tail end of the embryo, the opposite end of the primitive streak is marked by a dome of epiblast cells the primitive node (also known as Hensens node in chicks and the blastopore in amphibians).
Cells of the epiblast are held in a sheet by a variety of cellular junctions (gap junctions, tight junctions and adherens junctions). Signalling proteins (such as E-cadherin) released by cells in the region of the node induce changes in the surrounding epiblast cells that enable them to alter these junctions, loosening the adhesion to their neighbours and to activate their cytoskeletal systems. This enables epiblast cells to migrate toward, and into the primitive streak in what is one of the most defining processes of gastrulation. This process of internalisation or ‘emboly’ involves, not only coordinated movement of cells through the primitive streak to lie beneath the epiblast but also a transformation in cellular identity that results in the formation of endoderm, mesoderm and ectoderm, a change known as epithelial to mesenchymal transition (EMT) [10,11].
Early migrating epiblast cells that descend into the primitive streak in the process of emboly, displace the underlying hypoblast cells to progressively more lateral positions, ultimately replacing the hypoblast with a new layer, the definitive endoderm. Cells that existed in a more peripheral position in the epiblast have a delayed response to the nodal signalling (as they lie further along the concentration gradient of signalling proteins), when the these cells eventually enter the primitive streak they occupy a space between the recently formed endoderm and remaining overlying epiblast; during this process these are transformed into mesodermal cells. Those cells of the epiblast that fail to migrate, remain as a layer on the dorsal aspect of the embryo, the ectoderm.
In addition to individual cellular movements and the process of internalisation or emboly, another important aspect of gastrulation involves the coordinated movement of entire sheets of cells. Epiboly describes the morphogenetic process by which tissues expand and grow often by thinning out, a process that is crudely analogous to rolling out pastry. However, such a process alone results in a non-directional expansion, and so to ensure that the craniocaudal configuration of tissue growth is maintained another evolutionarily conserved process known as convergence and extension is required. The process of convergence and extension entails changes in cell shape and movement that lead to rearrangements of cells within a sheet such that a progressive extension occurs in a longitudinal direction with a concomitant narrowing in the transverse direction. The non-canonical Wnt pathway and the planar cell polarity (PCP) pathway are now known to be integral to this process [24]. During gastrulation, the down-regulation of cell-cell adhesion molecules, permitting cellular movement, combined with convergent extension growth, gradually transforms the circular symmetry of the bilaminar disc into the elongated post gastrulation embryo; one that has dorsoventral and left right and cranio-caudal orientation.
Neurenteric canal and notochord
The primitive node defines the upper limit of the primitive streak which, prior to its regression extends along the caudal half of the embryo. In amphibian embryos the node has been studies in detail. The dorsal lip of the node is the site of the Spemann-Mangold organiser, a centre that is of vital importance in co-ordinating the axial patterning events that constitute gastrulation. Wnt, VegT and BMP are three of the principle signalling pathways active in this site [33]. Cells from the node form a tube of cells, beneath the epiblast, that extends cranially (in the opposite direction to the primitive streak), constituting the notochordal process. As further growth of the notochord process takes place the primitive streak regresses caudally until it reaches the level of the cloacal membrane.
The notochord tube is temporarily incorporated into the endoderm forming the notochordal plate, once this happens there is a transient connection between the yolk sac and the amniotic cavity at the level of the primitive pit. This connection is the neurenteric canal. The notochord plate, subsequently separates from the endoderm to form a solid cord of cells that extends along the midline, limited by the bucopharyngeal membrane at the cranial end and the cloacal membrane and the caudal end. The notochord, via sonic hedgehog signalling (SHH) is essential in inducing folding of the overlying neural plate and in establishing vertebral segmentation [3].
The specific details of gastrulation vary between species and any extrapolation to the human model has to be interpreted with caution. Nonetheless the movement of cells, actively through cytoskeletal action or passively by displacement, the transformation of cells in response to this movement and changes in the topography of sheets of cells imposed by geometric changes seem common themes in all species studied so far.
Go to : 
HUMAN DISORDERS ATTRIBUTED TO ABNORMAL GASTRULATION
There are a number of human malformations and disorders whose etiology has been attributed to abnormal gastrulation, these include conjoined twinning, chordomas, malformations of caudal development and some of the spinal dysraphic entities (Table 1). From the paediatric neurosurgical perspective it is the dysraphic malformations that are most commonly encountered in clinical practice. Whilst much has been written on the presumed embryological basis of spinal dysraphism, and indeed most classifications of spinal dysraphism are based on these hypotheses, it is only in the defects of neural tube closure that there is strong supportive evidence from animal studies. Nonetheless, whilst acknowledging the limitations of current classifications and evidence for causation it is helpful to review some of the existing hypotheses in respect to spinal dysraphism since such developmentally based models of dysraphism can help the neurosurgeon in understanding the surgical anatomy of these complex lesions.
Table 1.
Congenital anomalies that have been attributed to disordered gastrulation
| Gastrulation event | Anomaly | Clinical consequence | Reference |
|---|---|---|---|
| Primitive streak | Duplication | Conjoined twinning | [33] |
| Notochord | Notochordal rests | Chordoma | [17] |
| Ecchordosis physaliphora | [18] | ||
| Tornwaldt cysts | [20] | ||
| Notochord | Notochord splitting | Split cord malformations | [13] |
| Neurenteric canal | Accessory neurenteric canal | Neurenteric cyst | [9] |
| Caudal mesoderm | Impaired migration | Caudal agenesis | [26,27] |
| Sirenomyelia | |||
| Caudal mesoderm | MNX1 mutation | Currarino syndrome | [32] |
![]()
As described above, at the end of gastrulation the three germ cell layers and the notochord have been established as a prelude to neural tube formation. It is reasonable to suppose that any dysraphic abnormality that has its origins in gastrulation might exhibit some or all of the following features : 1) evidence of involvement of other germ cell layers in addition to ectoderm; 2) the principal anatomical features of the dysraphism will be ventral to the neuraxis; and 3) coexisting anomalies of vertebral development commonly seen.
Spinal dysraphism
The spectrum of dysraphic malformations that presents to the paediatric neurosurgeon is broad moreover there are differences in natural history within the various subtypes and the mechanisms by which they result in neurological disability (dysplasia, mechanical tethering, ischaemia etc.) are also variable. Attempting to classify dysraphic conditions and ascribing an accurate diagnosis is therefore important, not only to guide management of the individual patient but also to ensure that clinical research is not confounded by the inclusion of disparate pathologies. The classification and nomenclature of spinal dysraphism has evolved, particularly in the past few decades, however there remain many instances where the embryological, radiological and clinical perspectives are difficult to reconcile.
Current classifications of spinal dysraphism are largely based on the presumed embryological stage at which spinal cord development is affected. Although this approach is attractive it is important that its limitations are recognised and the following points are acknowledged. Firstly, whilst animal models exist for open neural tube defects this is not so for the ‘closed dysraphisms’ making it difficult to test theories regarding aetiology. Secondly, with respect to normal gastrulation, the exact processes underlying this stage of development differ amongst animal models studied so far and so some caution needs to be exercised in making any extrapolation to abnormal gastrulation in humans. For example the very existence of the neurenteric canal (central to hypotheses of neurenteric cyst formation) in human embryos has been debated [25]. Thirdly, with the exception of rare instances (e.g., Currarino syndrome) extensive research has failed to identify consistent gene mutations in occult spinal dysraphism disorders. The mechanisms underlying the embryopathy in these cases remains largely unknown and it is likely that epigenetic and environmental factors may also play a role; the association between folic acid deficiency and open neural tube defects, and the relationship between maternal diabetes and caudal regression syndrome are strong evidence of such.
Neurenteric cyst
Neurenteric cysts (also known as enterogenous cysts, enteric cysts, archenteric cysts, bronchogenic cysts) are rare congenital cysts usually considered to be part of the dysraphic spectrum. The cyst wall has histological features that suggest an endodermal origin (e.g., mucin recreating cells), the cysts mostly occur ventral to the neuraxis and anomalies of vertebral development are frequent, hence these anomalies satisfy the criteria for gastrulation disorders suggested above.
During gastrulation the position of the neurenteric canal moves along the dorsal aspect of the embryo as the primitive streak regresses with concomitant formation of the notochord. Neurenteric cysts are considered by many to represent a remnant of an early endoderm/ectoderm interface at the site of the neurenteric canal. However, the extent of the neurenteric canal is limited by Hensen’s node rostrally and by the cloacal membrane caudally, an observation that has implications for the genesis and location of neurenteric cysts. Since the neurenteric canal is located at the inferior limit of the notochord, simple persistence of the neurenteric canal cannot explain the ‘dorsal enteric fistula’ characteristic of neurenteric cysts seen in clinical practice. It was to circumvent this paradox that Bremmer postulated the need for an accessory neurenteric cana [l1]. This concept has persisted yet there remains little experimental evidence for this supposition. If the origin of neurenteric cysts is related in some way to the neurenteric canal then their anatomical location should be confined by the rostrocaudal boundaries of the notochord defined above, and so should only found along a line extending form the sphenopalatine synchondrosis of the clivus to the upper sacrum. Indeed this is what appears to be the case, neurenteric cysts occur in the posterior fossa and along the spine but not in the supratentorial compartment and not in the sacral canal. The cervical spine (including the craniocervical junction) followed by the thoracic spine are the most common locations for neurenteric cysts [20]. Furthermore since gastrulation precedes folding of the neural plate, neurenteric cysts are characteristically found ventral to the spinal cord and whilst intramedullary neurenteric cysts do occur the majority are located in the intradural extra medullary compartment [18,28].
Neurenteric cysts, like split cord malformations are frequently associated with abnormalities of the paraxial mesoderm resulting in vertebral body malformations including abnormal segmentation (block vertebrae) and hemivertebrae in more than half of cases (Fig. 1).
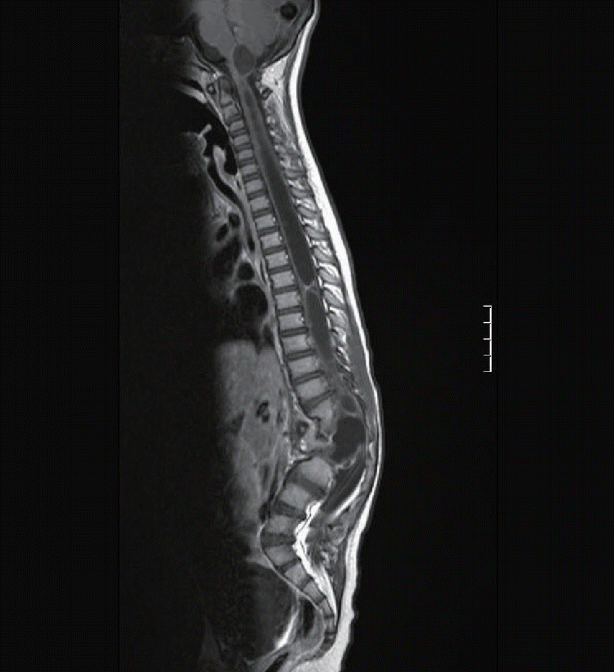
Split cord malformations
In 1992 Pang and colleagues [21] proposed the currently accepted nomenclature, type I and type II split cord malformation (SCM) for the conditions that were previously termed diastomatomyelia and diplomyelia respectively. Fifty years previously Bremer had hypothesised the existence of an accessory neurenteric canal to explain the origin of these malformations. Common to these hypotheses is the concept of an abnormal, or persistent, adhesion between the endoderm and ectoderm leading to an ‘accessory’ neurenteric canal. This adhesion, it is proposed, would disrupt notochord formation (by preventing the midline integration of mesodermal notochord progenitor cells) causing it to split, resulting in a duplication of the notochord and the subsequent induction of two hemicords. Pang, elaborated on the hypothesis in an attempt to embrace the two commonly recognised forms of SCM. He suggested that mesenchyme would condense around the endodermal-ectodermal adhesion to form an endomesenchymal tract. The inclusion (or exclusion) of primitive mesenchymal and meningeal progenitor cells into this endomesenchymal tract, and their subsequent maturation into bone and dura would determine whether the two hemicords came to lie within adjacent dural sacs, separated by an intervening bony spur (type I SCM), or whether they ended up in a single dural tube, separated by only fibrous remnants of the accessory canal (type II SCM) (Fig. 2).
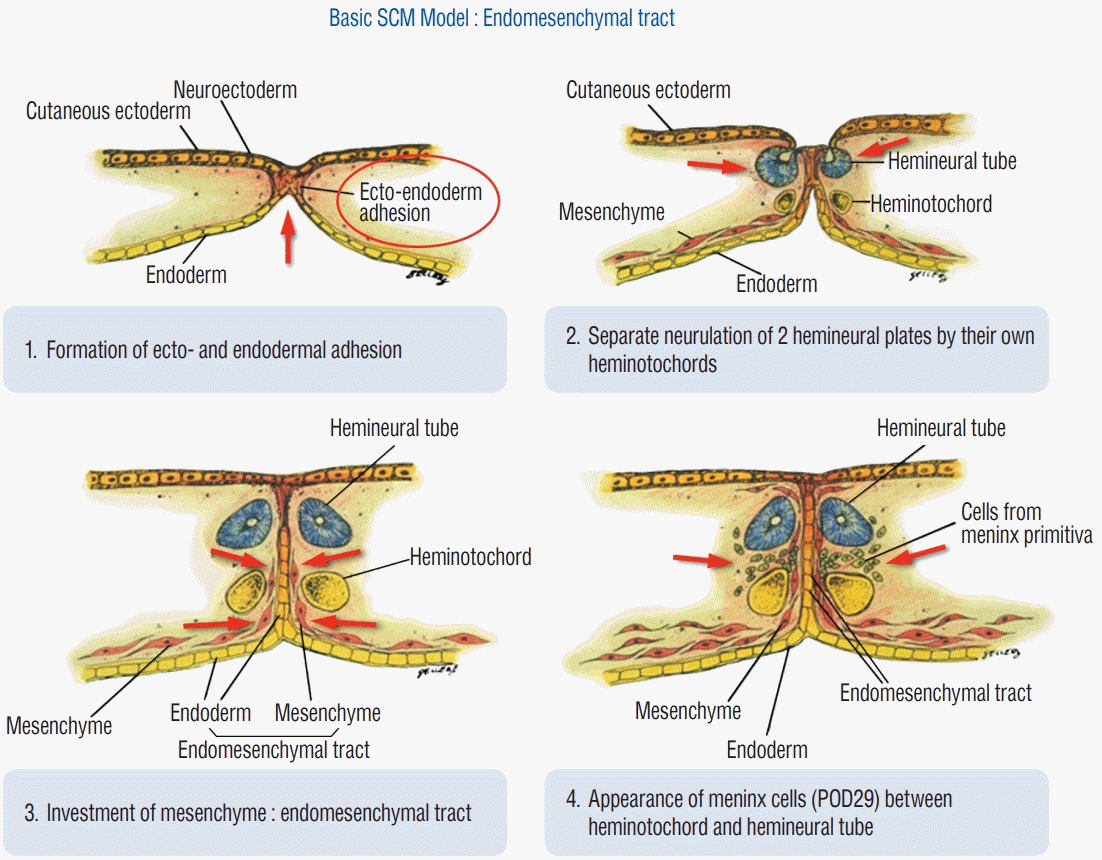 | Fig. 2.Diagrams illustrating the putative endomesenchymal tract and its relationship to the formation of split cord malformations. Adapted from Pang et al.[21]. SCM : split cord malformation, POD : post ovulation day.
|
Although the two SCM types are quite discrete, the two can occur in the same individual, so called ‘composite SCMs’ [30]. What determines the formation type I rather than type II is unknown, though Pang has proposed that this may relate to timing of the embryonic insult based on the observation that type II SCMs are typically seen in the cervicothoracic region whereas type I are more commonly encountered in the low thoracic and lumbar levels. Vertebral malformations (segmentation anomalies of fusion or form) as well as dysplastic laminae are more commonly seen in type I SCM (Fig. 3).
Although an accessory neurenteric canal has not been observed in an animal model and its occurrence remains speculative, Emura et al. [6,7] sought to create an experimental model of accessory neurenteric canal by passing a fine needle from the neural plate to archenteron to create a fistula across the an amphibian embryo (Cyanopus pyrrhogaster). On subsequent histological analysis they observed partial duplications of the spinal cord and notochord [6,7].
Notochordal anomalies
The notochord has a major role in the reorganisation of paraxial mesoderm to form the vertebral column. The nucleus pulposus is considered to be a persisting remnant of the notochord. A number of rare pathological entities are thought to arise from cells of notochordal origin [26]. These include chordomas, rare malignant tumours of the vertebral column seen in children as well as adults. These tumours demonstrate a predilection for the clival and sacral regions and this correlates with the extent of the notochordal derivatives, sphenoid synchondrosis to sacrum.
Ecchordosis physaliphora (also known as benign notochordal cell tumour, BNCT) is an uncommon lesion derived from notochordal remants. The commonest site is the clivus, often identified as incidental findings on magnetic resonance imaging (MRI) where they appear as non enhancing T1 hypointense, T2 hyperintense cyst-like structures [17]. The differential diagnosis includes chordoma, indeed the two entities share some histological and immunohistochemical features, and it has been suggested that BNCT might even represent a precursor of chordoma. The majority are asymptomatic and do not require treatment although they can cause symptoms via mass effect or bone erosion with cerebrospinal fluid fistula formation [4].
Tornwaldt cysts are benign developmental cysts that typically occur in the posterior nasopharynx, between the longus capitis muscles. These, too are thought to result from persistent remnants of a nasopharyngeal (endodermal) to notochord connection [27].
Caudal agenesis (and sirenomyelia)
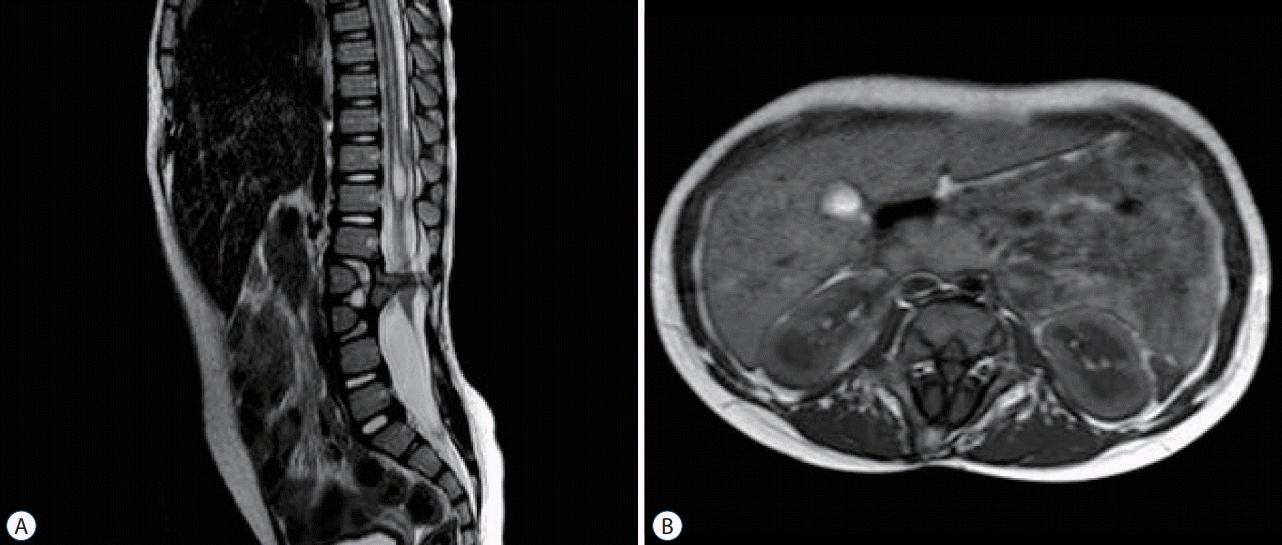
The radiological features of the caudal regression syndrome are characteristic and comprise a high, abrupt termination of the spinal cord (corresponding to the S1 level), dysplasia of the lower sacral segments and anomalies of the renal and genitourinary tract (Fig. 4). The appearances of the spinal cord suggest a complete absence of those structures deemed to arise from secondary neurulation; conus medullaris, filum terminale and elements of the caudal equina. The coexistence of vertebral and hindgut malformation raises the possibility that causal agenesis could have its origins in gastrualtion. Impaired caudal mesoderm differentiation is thought to have an etiological role, and there is experimental evidence to support this. The transcription factor Brachyury is known to be essential in caudal mesoderm development and knockdown mouse models in which Brachyury expression is impaired, exhibit spinal and genitourinary malformations reminiscent of human caudal regression syndrome [22]. However, in the human no clear genetic predisposition has been identified. An association between caudal regression and maternal diabetes is well known suggesting a role for environmental factors, however less than one quarter of infants with caudal regression are born to diabetic mothers.
Abnormal migration of cells that constitute the caudal mesoderm has also been postulated as a mechanism underlying a spectrum of disorders that includes caudal regression syndrome and sirenomyelia. Experimental evidence to support this hypothesis has not been forthcoming and there appear to be other mechanisms, for example vascular (abnormal umbilical vasculature is common to both conditions) that now seem to provide more promising explanations for these disorders [10,29]. Retinoic acid administration to mice has also been shown to cause patterns of caudal dysgenesis that include spinal dysraphism and disorders of the lower genitourinary tract as well as sirenomyelia [19].
Caudal genitourinary malformations and spinal dysraphism
Spinal dysraphism commonly occurs in conjunction with urogenital and anorectal and malformations for example cloacal extrophy and the urorectal septal malformations sequence (URSMS). In a recent audit 34/35 children with cloacal extrophy had MRI evidence of spinal dysraphism (unpublished data; Fig. 5).
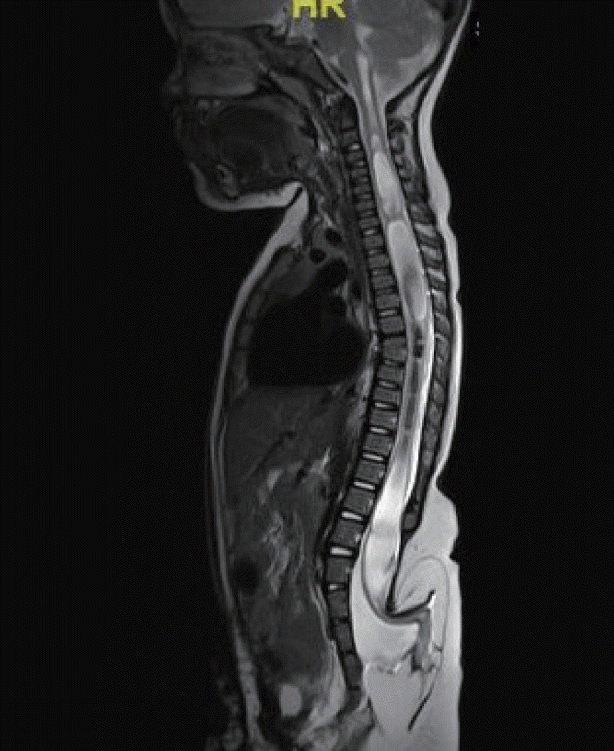
Failure of the urorecral septum to fuse with the cloacal membrane, which in humans occurs between 4–6 weeks, was initially proposed as a mechanism to account for the URSMS [9]. However the extent of the anomalies that typically coexist in this group of disorders seems incompatible with such a specific defect leading to speculation that an earlier embryological insult must be responsible. Genes including SHH, HOX and PAX that are essential for endoderm-mesoderm signalling and for mesodermal cell movements in gastrulation are also expressed in the developing urorectal septum but it remains unknown mutations in such genes is directly responsible for these conditions. Teratogens such as retinoic acid (through down regulation of SHH) and etretinate have been shown in animal models to reduce cell proliferation in the caudal mesoderm and result in urogenital malformations reminiscent of URSMs [8,14].
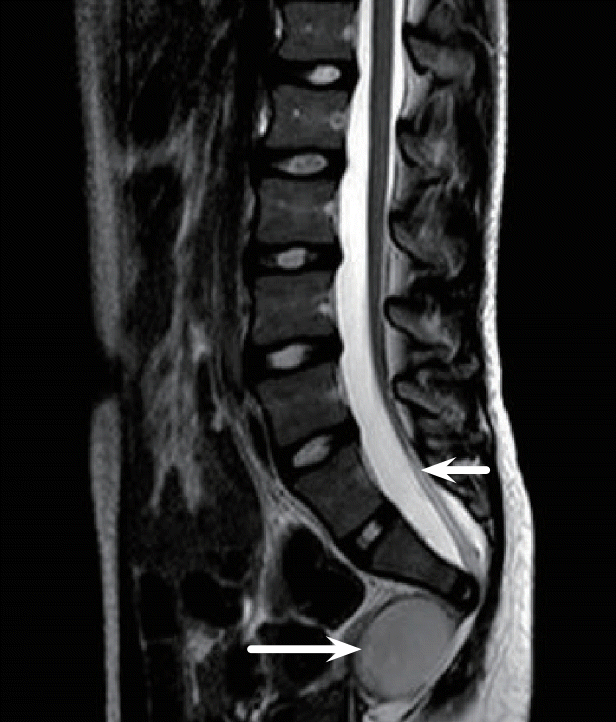
Currarino syndrome
The Currarino triad of anorectal anomaly, sacral dysgenesis and presacral mass is unusual amongst the caudal malformation syndromes in that a gene mutation has been identified (MNX1). Currarino syndrome follows an autosomal dominant pattern of inheritance though sporadic cases are frequent. A mutation in the MNX1 gene is found in 90% of familial cases but in only half of sporadic cases. Many different sites of mutation have been described within the MNX1 gene and the correlation between genotype and phenotype is inconsistent. Penetrance is low and many cases are discovered incidentally or have minimal symptoms [15]. The nature of the presacral mass is quite variable and includes cystic lesions such anterior meningocele and dermoid cyst as well as solid tumours that are usually benign hamartomas or mature teratomas [2]. Although spinal dysraphism is not an obligate diagnostic feature of the syndrome, abnormaities of the terminal spinal cord, in particular spinal lipoma, occurs in more than two thirds of cases [2,16] (Fig. 6). Currarino triad therefore satisfies our criteria for a lesion with potential origins in gastrulation, combining dysraphic, vertebral (sacral dysgenesis) and mesodermal (presacral mass) involvement. Dias and Azizkhan described a case of Currarino with split cord malformation, citing this as evidence for a common embryogenic origin due to failed dorsoventral separation of the caudal eminence from the hindgut endoderm toward the end of the gastrulation period [5]. Gegg et al. [13] reported neural elements within a presacral teratoma and suggested that this also indicated an origin in gastrulation. Beyond such case reports there is little evidence to link Currarino to a failure in gastrulation.
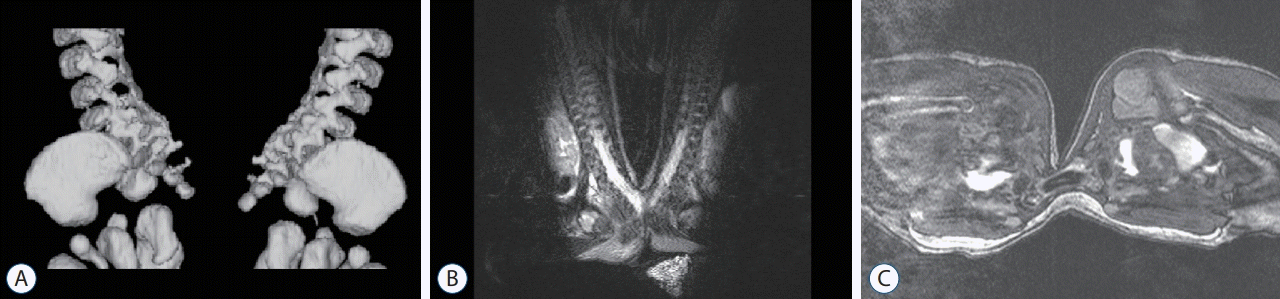
Twinning
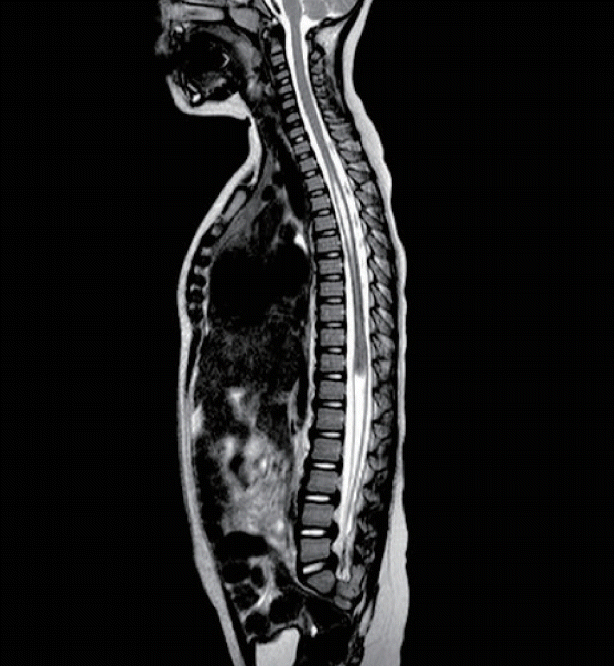
Monzygotic twins arise from a single fertilisation and are classified according to the partition of the placental and embryonic membranes. The type of twining is dictated by the precise timing of the separation. An early duplication at the morula or blastocyst stage that results in a pair of embryos that either share (monochorionic), or have separate (dichorionic) membranes but will have separate embryonic (amniotic) membranes. Rarely, duplication of the embryo occurs slightly later, at the onset of gastrulation with the formation of two separate primitive streaks. By this time the epiblast and amniotic cavity have already formed and so the twins are not only monochorionic but also share an amniotic cavity (monoamniotic). Any later attempt to separate may result in incomplete separation resulting in twins that are not only monochorionic, monoamniotic twins but are conjoined. Approximately 30% of conjoined twins are joined at the rump (pyopagus) or pelvic outlet (ischiopagus) and these may also have coexisting fusions of the terminal spinal cord segments (Fig. 7). Vertebral anomalies of fusion or form are seen in up to 75% cases [12].
Go to :
SUMMARY
The understanding of gastrulation has been transformed by the advent of molecular and cellular biology techniques. Much is now known of the interaction between evolutionarily conserved genes, signalling pathways and cellular activity in the primitive embryo. In addition to beginning to explain normal development, genetic manipulations in animal studies have begun to shed light on the mechanisms underlying some of the congenital malformations familiar in paediatric neurosurgical practice. Many of the theories of causation of developmental abnormalities of the spine and spinal cord have, and will continue to be challenged by these advances. Whilst there appear to be shared principles underlying gastrulation between different species there are important differences and results from animal studies cannot be simply extrapolated to human counterparts. With the exception of open neural tube defects animal models for spinal dysraphism do not exist, and in the majority of cases consistent genetic mutations have not been identified. Although classifying spinal dysraphisms according to developmental stage has some advantages in clinical practice, it is important that we use such classifications with caution and remain open to modifying these in the light of new discoveries in developmental biology. As exemplified by Prof. Wang and colleagues in Seoul the next generation of paediatric neurosurgeons need to embrace collaborative translational research in order to facilitate these advances.
Go to :
 XML Download
XML Download