

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Brain tumors are the most common solid tumors during childhood. In adults, mutations which are observed in most tumors are caused by external insults [115]. In contrast, a large subset of mutations in pediatric brain tumors will originate in the germline as part of cancer predisposition syndrome (CPS). According to Knudson’s “two hit” hypothesis, the germline mutation results in a first hit, allowing for a higher chance of a single somatic “second” hit to cause cancer. While this model may not explain the genetic etiology of all heritable cancers, it has been a guiding principle for cancer susceptibility and pathogenesis [17].
At least 10% of children with cancer are primarily affected by a CPS [76,80,115]. However, newer estimates are much higher [17,56]. Recognition of a CPS is crucial for patients and their family members. Specific germline mutations confer different risk for tumor response and survival as well as offer mutational derived therapies. Moreover, once discovered, surveying patients with CPS enables early detection and has proven to improve survival [86,109].
Specific brain tumor types are associated with higher prevalence of germline mutations. Atypical teratoid/rhabdoid tumors during early childhood carries up to 35% risk of belonging to the Rhabdoid predisposition syndrome, caused by germline SMRCB1 or rarely SMARCA4 mutations [26]. On the other hand, each CPS has a unique group of brain tumors which are specific to the germline mutation. For example, patients with Gorlin syndrome harbor an increased risk for early onset sonic hedgehog medulloblastoma (MBSHH). These MBSHH confer better survival than other medulloblastoma at this age.
Pediatric high grade gliomas (PHGG) are the deadliest group of childhood brain tumors. In this review, we will focus on PHGG and describe three of the more prominent syndromes in these cancers–Li Fraumeni syndrome (LFS), constitutional mismatch repair deficiency and neurofibromatosis 1. Table 1 summarizes the major features of those three CPS.
LI FRAUMENI SYNDROME
LFS is an autosomal dominant CPS and one of the hallmarks of such syndromes in children and adults. LFS is characterized by high frequency of malignancies in multiple organs and lack of other clinical features [71]. Estimates of prevalence are 1 in 5000–20000 individuals [34,59], though recently it was suggested to be an underestimation [4].
LFS has classical clinical criteria (Table 2). However, following the identification of germline mutations in the TP53 as the syndrome’s etiology [71], more cases that were not fitting the classical LFS criteria were found [34,110]. Hence, the clinical criteria have been revised several times, to the recent more comprehensive “revised Chompret criteria” [16,104]. These criteria are therefore merely a guidance for individuals who should be referred for a genetic diagnosis, though not all of the families fitting the clinical criteria harbor detectable germline p53 mutations [34,69,70,78].
Cancer spectrum
The penetrance of cancer is high, yet it is highly variable. The lifetime risk of developing at least one cancer is approximately 75% in men and 93–100% in women, and up to 41% of children will have cancer by age 18 years [16].
The cancer spectrum is vast and includes sarcomas, premenopausal breast cancer, adrenocortical carcinoma (ACC), brain tumors, hematologic malignancies and others. Brain tumors are the second most common malignancies in children with LFS following ACC, as 26% of childhood tumors are in the central nervous system (CNS), while only 13% of adult tumors in LFS are in this location [16,67]. The median age of onset of brain tumors in LFS is 16 years [78], compared to 57 years in the general population.
While malignant gliomas are the most common brain tumor in LFS, they tend to occur during late childhood and adulthood. In the younger age group, choroid plexus carcinomas (CPC) and Sonic MBSHH are more prominent [68]. The prevalence of LFS among patients with CPC is extremely high : 36–50% of patients with this tumor harbor TP53 germline mutation [36]. CPC is considered an LFS-defining tumor which obligates genetic testing to every patient with the tumor, regardless of family history [103]. Similarly, half of children with SHH/TP53-mutated medulloblastomas harbor germline mutations in TP53 [116], and should be referred to genetic counselling as well. Tumors of astrocytic origin in the context of LFS include mostly high grade gliomas but lower grade gliomas have been observed [10,16].
Biological considerations
TP53 is a tumor suppressor gene. The protein it encodes (p53) upregulates the transcription of target genes involved in cell cycle arrest, DNA repair, apoptosis and senescence, in response to DNA damage [41]. The TP53 gene is located on chromosome 17p13 and more than 250 different germline alterations of it have been reported. While the genotypic : phenotypic correlations are not fully understood, most mutations in brain tumors reside within the DNA binding domain [78]. Brain tumors seem to cluster in certain families with LFS, possibly due to additional modifying genes [55]. Somatic inactivation of TP53 remains one of the most frequent genetic change identified in human cancer [6,50] including childhood glioblastoma [79].
The biology of the glioma itself in individuals with LFS may differ. Watanabe et al. [111] reported a rare type of IDH1 (R132C) in LFS gliomas. Since IDH1 mutations are common in secondary glioblastomas of young adults which progress from lower-grade tumors [21,82], these observations may be important for the management of these patients.
Clinical implications
Diagnosis
Histologically, CNS tumors associated with TP53 mutations are identical to their sporadic counterparts. Since TP53 mutations exist in up to 50% of PHGG, positive tumoral immunostain or even somatic mutation in TP53 does not correlate with germline mutations. Since LFS is a highly penetrant syndrome, clinical-familial diagnostic criteria are the main indication to search for the presence of TP53 germline mutation, which is sufficient for diagnosis [34,94,104].
Management
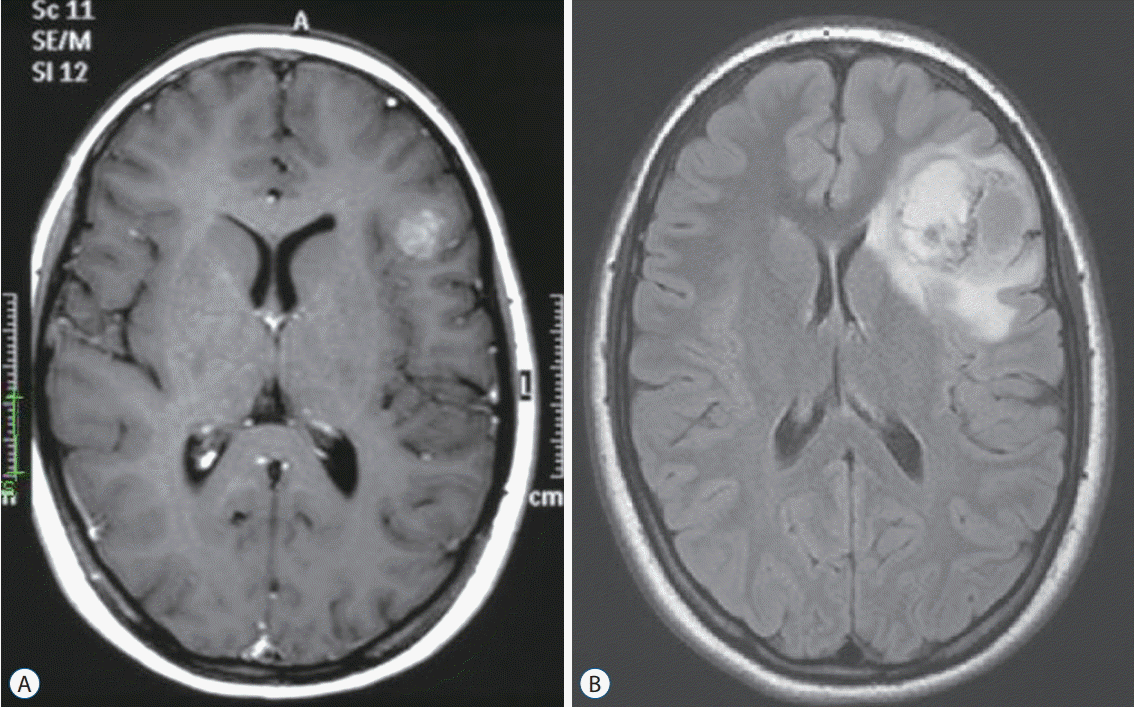
TP53 mutations were proven to be a negative prognostic factor in several tumor types, including CPC [103], MB [100,116], and PHGG [27,83]. Therefore, with current treatment approaches, late detection of LFS gliomas when these are already PHGG may not be sufficient for curative intent. Since transformation of lower grade gliomas to PHGG have been reported [13,102] (Fig. 1), early detection and resection of a low grade lesion may offer improved survival for LFS individuals.
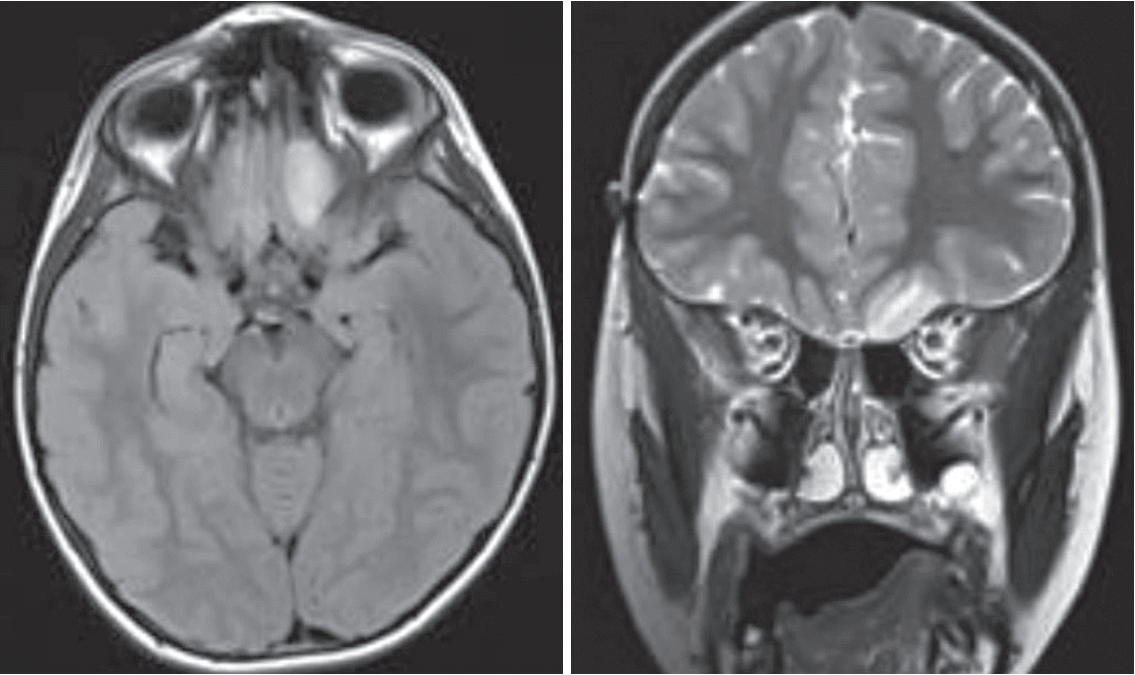
Villani et al. [109] demonstrated a survival advantage in patients with LFS undergoing intense tumor surveillance. Forty tumors were detected in 19 of 59 patients on surveillance, including gliomas that were detected with brain magnetic resonance imaging (MRI). Furthermore, 25 of 40 tumors found on the surveillance protocol were low grade or premalignant at the time of detection, suggesting that early detection through surveillance may identify lesions before malignant transformation. Five-year overall survival was 88.8% versus 59.6% in individuals not undergoing surveillance. Other studies have recently confirmed improved clinical outcomes for TP53 mutation carriers with intensive screening [10,14,92]. Complete resection of lower grade gliomas from LFS patients (Fig. 2) may improve survival by prevention of the transformation to PHGG.
Table 3 summarizes consensus recommendations made by an international expert group concerning surveillance of LFS [57]. The lifelong brain tumor risk justifies dedicated annual brain MRI. Annual whole body MRI that is recommended for solid tumor surveillance, cannot replace dedicated CNS imaging, as was proven by a recent meta-analysis [10], and ideally should alternate with the brain MRI every 6 months.
Treatment
Currently, there are no TP53 specific therapies and the prognosis for LFS-associated HGG remains poor [102]. Since p53 plays a key role in response to DNA damage, the risk of secondary malignancies including PHGG post genotoxic damage from chemoradiation is high. Increased risk of therapy associated secondary malignancies in the radiation field was reported in number of cohorts and case reports [16,42,44,63,65,96]. However, currently, taken the challenges of treating PHGG, there are no successful alternative treatment strategies for cancer in the context of LFS.
CONSTITUTIONAL MISMATCH REPAIR DEFICIENCY (CMMRD)
CMMRD is a childhood cancer syndrome caused by biallelic mutations in the mismatch repair pathway [113]. Monoallelic mutations in MMR genes results in a CPS termed Lynch syndrome. This autosomal dominant syndrome presents with primarily gastrointestinal and genitourinary malignancies in mid to late adulthood. In contrast, biallelic mutations in the MMR genes causes complete loss of MMR ability in all cells resulting in CMMRD, an autosomal recessive syndrome with vast spectrum of malignancies and grave prognosis during childhood.
Previously, CMMRD was also termed as brain tumor-polyposis syndrome-1, biallelic MMRD (BMMRD), or Turcot syndrome type 1. It is important to differentiate CMMRD from Turcot type 2 (familial adenomatosis polyposis, FAP) which is an autosomal dominant syndrome caused by germline mutations in the adenomatous polyposis coli (APC) gene. FAP has similar characteristics of numerous colonic adenomas and progression to colorectal carcinoma. However, brain tumors are rare and are almost exclusively related to medulloblastoma during childhood and rarely astrocytoma, ependymoma, and pinealoblastoma [108].
Biological considerations
The mismatch repair system is one of the major DNA repair pathways in humans and is composed of several genes including MSH2, MSH6, MLH1, and PMS2 [112]. Its primary function is to correct errors that arise during DNA replication. Hence, mutations in MMR genes lead to accumulation of somatic mutations in each cell division and can lead to hypermutant cancer. The two major types of mutations resulting from lack of MMR are point mutations (single nucleotide variations) and microsatellite instability (MSI) in which mutations repetitive sequences (microsatellites) are not adequately repaired.
Recently, other components of the replication repair machinery have been reported to be associate with similar clinical and biological presentation and cancer hypermutations. These included mutations in MSH3 [1], deletions of the EPCAM gene, located just upstream of MSH2 [64] and mutations in DNA polymerases epsilon and delta 1 (POLE, POLD1) [29,77].
Since most mutations in the MMR genes result in lack of protein expression, there is no clear genotype phenotype correlation. However, in contrast to Lynch syndrome where MSH2 and MLH1 are the most common genes affected, PMS2 mutations are most frequent in CMMRD, followed by MSH6 [113].
Clinical implications
CMMRD patients frequently present with physical features, the most common being café au lait spots or other hyper- and hypopigmented skin alterations. Not infrequently they are misdiagnosed as NF1. Other features diagnostic for NF1 may be apparent as well but are far less frequent, including neurofibromas, freckling, Lisch nodules and others [113,114]. Other physical finding that can sometime be found in CMMRD are venous anomalies, pilomatricomas (benign skin lesions), agenesis of the corpus callosum [7], and decreased levels of immunoglobulins IgG2/4 and IgA [113]. As it is an autosomal recessive syndrome, consanguinity is a common feature, however none of these clinical and familial features is obligatory [60,113].
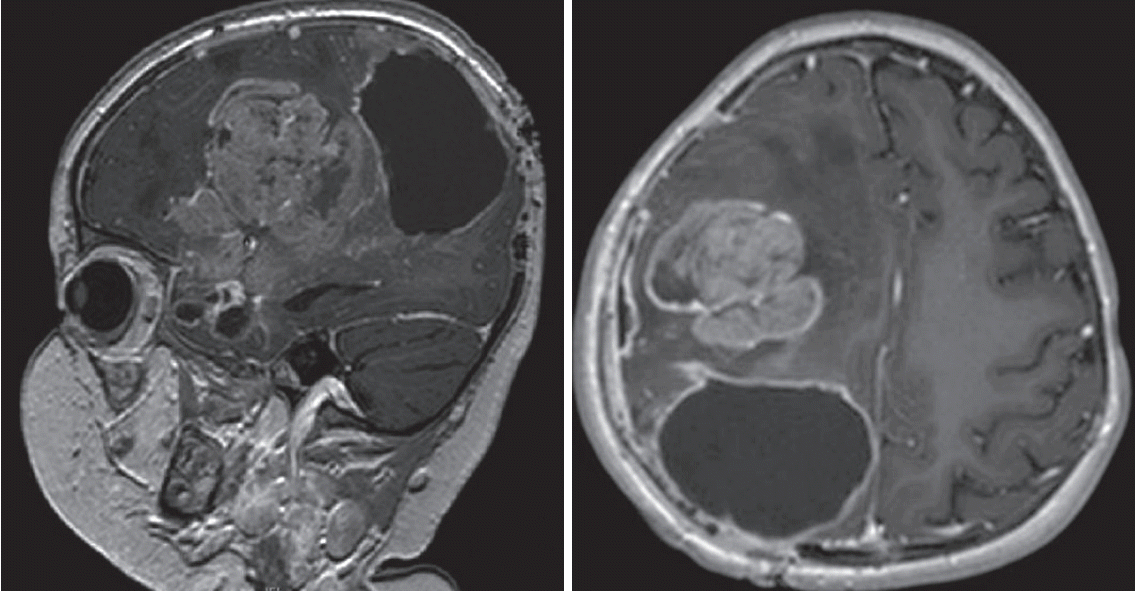
Penetrance is extremely high reaching more than 90% at age 20, hence almost all patients will have cancer as children. In fact, since mutations are so abundant in the setting of deficient corrective mechanisms, most individuals will have more than one tumor, which can occur metachronously or synchronously (Fig. 3).
PHGG constitute the most prevalent brain tumors in patients with MMR mutations, although medulloblastoma, supratentorial primitive neuroectodermal tumors and low grade gliomas have also been reported. Median age at diagnosis of brain is 9–10.3 years, nevertheless they were observed since infancy [9,60,113]. Other malignancies include hematological malignancies (mainly T-lymphoblastic lymphoma), early onset of colorectal cancers and virtually every organ can be affected.
Diagnosis
CMMRD should be suspected in children and adolescents with PHGG (or other malignancy) that have café au-lait macules, a history of Lynch syndrome in the family, or a sibling with childhood cancer. However, other features can occur as mentioned and thus high index of suspicion is required [3,60,101]. Since PHGG are uncommon in neurofibromatosis-1 (NF-1), every child with “known” NF1 and a malignant tumor including PHGG during childhood should be investigated for CMMRD. Immunohistochemistry showing loss of one of the MMR proteins in both malignant and normal cells in the biopsy specimen is both sensitive and specific method for detection of CMMRD. This assay is available in most pathology laboratories as part of the routine workup of colon cancers in adults [9]. It will also guide target-gene mutation analysis for the corresponding mutated gene. However, some missense mutations will result in retained staining of the protein, hence a positive stain does not preclude a diagnosis of CMMRD [60]. In contrast, MSI which is extremely useful tool in Lynch syndrome cancers, is not high in CMMRD cancers and especially in PHGG. This tool therefore, should not be used as it can cause false negative testing and mismanagement of patients and tumors.
High tumor mutational burden, which is rare in childhood cancers, has been described to be extremely specific to CMMRD [5,45,99]. This vast numbers of mutations form a “signature” that is deeply engraved on the genome. In a recent study, this characteristic has been validated, as hypermutant childhood cancers were almost invariantly caused by replication repair deficiency and mutational burden and their signatures could be traced to the germline [19]. Finding of such hyper mutant primary tumor should therefore be followed by testing for CMMRD.
The final confirmation of the diagnosis of CMMRD should come from the determination of the causative biallelic mutations of the patient. However, mutation analysis is frequently difficult in case of PMS2 due to pseudogenes and variance of unknown significance in others. Therefore, a combination of clinical parameters (Table 4) and the above functional assays may be required.
Management
Once diagnosed, surveillance is crucial, as most children with CMMRD will be affected with cancer. A consensus surveillance protocol was established and published by the American Academy of Cancer Research [101] and other groups [25], and it’s summery is depicted in Table 3. Since CNS tumors are observed from infancy, imaging is recommended as soon as the diagnosis is done, and should be done every 6 months. MRI is the gold standard and ultrasonographic assessment cannot replace it, even in the setting of open fontanelle.
Treatment
There is currently no evidence of extensive toxicity of chemoradiation in CMMRD patients [60]. In contrast to other DNA damage repair deficiencies, repair of external insults is maintained as MMR is responsible for mistakes which occur during replication only. However, there is known tumor resistance to several common chemotherapeutic agents which require adequate mismatch repair to exert their tumor damage. These include Temozolomide, which is vastly used for PHGG treatment [54].
Tumors with high mutational load have increased formation of neo-antigen, which may serve as targets for the immune system [24,61,88]. This observation proved to be clinically significant in CMMRD PHGG as immune checkpoint inhibition was shown to have significant effect in prolonging survival for two patients with CMMRD recurrent glioblastoma [15]. It also held true in a patient with hypermutant glioblastoma multiforme (GBM) and POLE germline [49], and in non-CNS cancers [61,88,91]. Recently the Food and Drug Administration (FDA) approved the PD-1 inhibitor pembrolizumab for the treatment of mismatch-repair–deficient cancers [62]. These approaches and in combinations with others offer hope for patients with CMMRD PHGG and active pursue of international clinical trial is recommended.
NEUROFIBROMATOSIS TYPE 1 (NF1)
NF1 (von Recklinghausen disease) is the most common CPS, with an incidence of 1 : 2000–1 : 5000. As most CPS it is autosomal dominant, however the occurrence of cancers is only a part of this clinical syndrome. Approximately half of the cases occur de novo with no familial history [31]. The clinical diagnosis requires the fulfilment of at least two of the criteria (Table 5), however there are other possible manifestations, including macrocephaly, learning disabilities, vasculopathies, scoliosis among others. Penetrance is complete with some degree of clinical manifestations in every individual harboring a mutation [48].
Biological considerations
NF1 gene is a tumor suppressor gene located on chromosome 17q11.2. It encodes the protein Neurofibromin, a GTPaseactivating protein (GAPs) that inhibits the RAS oncogene by transforming GTP-RAS to GDP-RAS. RAS is an important activator of various signaling pathways, including the MAPK (RAF-MEK-ERK) and the PI3K/AKT/mTOR pathways [93]. As a consequence, a malfunction of its inhibitor results in increased proliferation and tumorigenesis. Importantly, since inhibitors exist for both pathways, NF1 mutations may be targetable for therapy.
Clinical implications
Most NF1 tumors are of benign nature. However, NF1 patients are at risk for malignant tumors including peripheral nerve sheath tumors (MPNST), PHGG and juvenile myelomonocytic leukemia (JMML) among others [48]. As for NF1 gliomas, these should be differentiated from foci of abnormal signal intensity (FASI), also termed UBOs (unidentified bright objects). These multiple, non-enhancing, small areas without mass effect or edema are benign, and are found in 70% of NF1 pediatric cases [52].
The most common CNS neoplasia in NF1 is optic pathway glioma (OPG), that tend to arise in infancy and affect approximately 15–20% of individuals with NF1 [2]. These OPGs are usually pilocytic or pilomyxoid astrocytomas. They can involve all parts of the optic tract; however, bilateral involvement of the optic nerves is highly suggestive of NF1 [90].
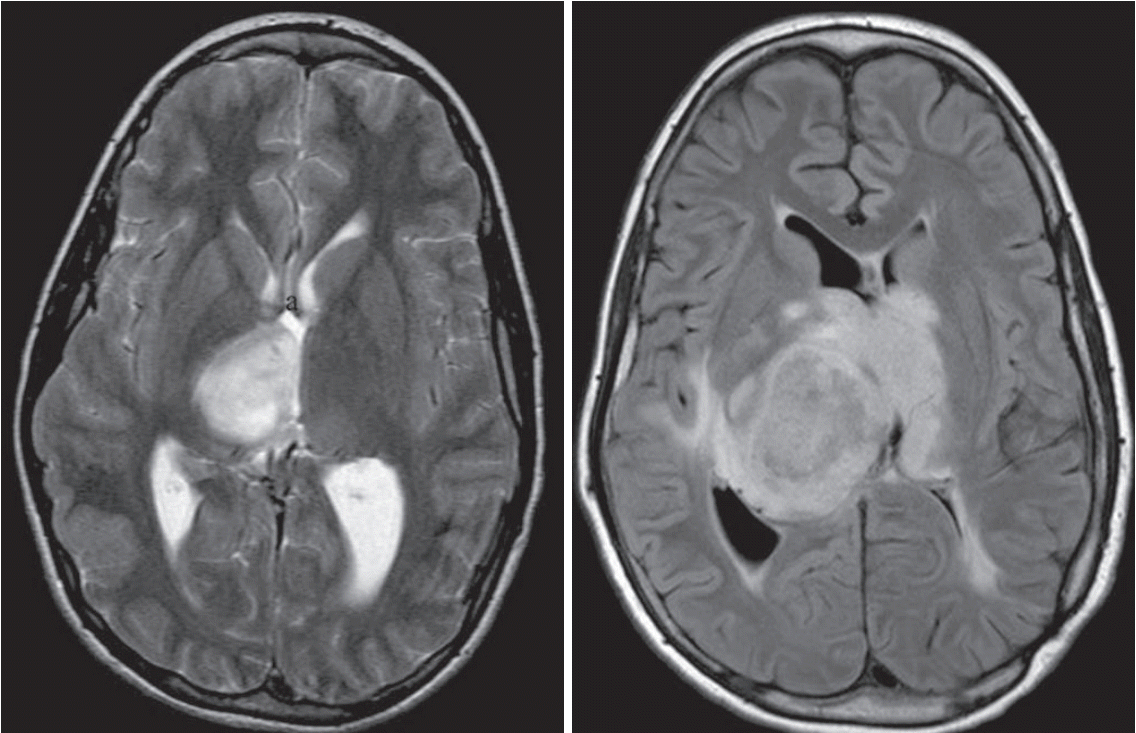
The rate of higher grade brain malignancies in NF1 is much lower. However, as shown in Fig. 4, this entity should not be underestimated [38,73,89,107]. Molecularly, NF1-PHGG share the same molecular abnormalities as non-syndromic patients, including secondary TP53 mutations and CDKN2A/p16 deletions [39]. Similarly, NF1 is one of the most frequently mutated somatic genes in sporadic glioblastoma [20,58,102]. It is still unclear whether NF1-PHGG are secondary gliomas arising from low grade lesions or can be primary PHGG [46,102].
Diagnosis
Although NF1 is traditionally diagnosed clinically, most of the features develop gradually and do not necessarily appear in the first months of life. Furthermore, other conditions with NF1 stigmata are known and include Legius syndrome and other RASopathies. Importantly, CMMRD (see above) can mimic NF-1 and the current expert consensus state that in the case of high-grade tumors including PHGG in a child with NF1, a genetic testing should be performed [30]. Genetic testing is thereby currently the standard of care. It is indicated for familial reasons, as well as for insuring correct NF1 diagnosis.
Management
Although the rate of optic gliomas is high, the role of surveillance neuroimaging in asymptomatic children with NF1 is still controversial [32,37,53,66,74]. Furthermore, since PHGG is uncommon in NF1, surveillance by imaging cannot be recommended [30,43]. Close monitoring with repeated ophthalmologic examinations and physical examination is the standard of care, and should include neurological examination and signs of endocrine malfunction [30,37,43,66,81] (Table 3). Families should be informed about the clinical warning signs of brain tumors and any evolving signs or symptoms should prompt investigations.
Treatment
In the case of imaging progression in conjunction with symptoms, medical therapy is often the modality of choice for low grade gliomas. Radiotherapy is not recommended due to numerous reports of complications specifically in the NF1 population. These sequela includes secondary malignancies and vascular complications, namely stroke [37,75,89]. Radiation as the cause of malignant transformation was emphasised in a study by Sharif et al. [95], where the relative risk for developing a secondary malignancy was 3.04 in NF1 patients treated by radiotherapy, compared with NF1 patients who did not receive radiation.
The treatment of NF1 PHGG is similar to sporadic cases. Although some indications exist that prognosis may be better than sporadic PHGG and prolonged disease is commonly reported, cure is not common [18,22,40,46,98,105,106].
MEK inhibitors, which have shown success in NF1 patients with low grade glioma [11], may offer improved outcome either alone or in combination with other therapies for NF1-PHGG.
OTHER CANCER PREDISPOSITION SY NDROMES
Other syndromes have been also associated less commonly with malignant gliomas. It is important to distinguish case reports from real increased risk for PHGG. Such CPS where there is no current data of increased risk include BRCA [28,33], tuberous sclerosis [87], multiple enchondromatosis [85], Fanconi anemia [23], Beckwith Widemann syndrome and more [12]. Adult syndromes can cause HGG but are rare in childhood. Familial melanoma astrocytoma is a CPS caused by inactivating germline alteration of the CDKN2A tumor suppressor gene. Individuals can develop both melanomas and astrocytomas (predominantly GBM), and occasionally other nervous-system neoplasms including peripheral nerve sheath tumors and meningiomas [51,84].
All of the aforementioned are cancer predisposition syndromes with a known single locus etiology. However, there are numerous cases in which we encounter a patient with a rich familial history, but none of the known germline mutations are found. Moreover, studies have shown families with an aggregation of gliomas, with up to a three-fold increased risk of glioma among close relatives [72]. A large study that included 5088 relatives of 639 probands diagnosed with a glioma under age 65 years, showed that such “familial glioma” are probably a result of multigenic action, and may involve unknown environmental exposures [4]. GLIOGENE is an international consortium which was formed in order to collect such non-syndromic glioma families, and identify new important genomic loci. Linkage studies have suggested linkage on chromosome 17q [47,97] but it is yet to be determined whether there is a clear disease causing mechanism. Exome sequencing of families in this consortium identified protection of telomerase protein 1 (POT1), which has also been implicated in melanomas, as potential glioma causing gene [8]. All of these may not cause or be associated with increased risk of PHGG and be more relevant to adults.
SUMMARY AND RECOMMENDATIONS
This review outlined the important role of several CPS in PHGG. Knowledge of specific CPS may alter the management of the cancer, avoid unnecessary treatment and offer novel targeted therapies which may improve survival for these patients. Furthermore, recognition of CPS may affect survival for other family members of children affected by PHGG.
As a result, when encountered with a PHGG, the physician should consider the possibility of CPS. Features of LFS, CMMRD and NF1 as mentioned above should raise a suspicion. However, even if their absence, other “universal criteria” are suggestive of a CPS [35]. These include any child with more than one primary tumor, a known CPS, or cancers in young family members, and should prompt referral to genetic testing.
If any of the above indications exist for suspecting a CPS, genetic counselling must be offered to the patient and their parents prior to performing mutation analysis. Psychological support should be offered, as identifying CPS has important implications not only to the proband but to his whole family, which needs to be genetically consulted as well.
Finally, consulting experts in the field of CPS can improve the management of the patient with CPS related PHGG and guide implementation of surveillance, preventive and potential therapies for other family members. In an era of precision medicine, molecular based therapies must be conjoined with thorough understanding of genetic causes of cancers, especially in children affected by PHGG.
 XML Download
XML Download