

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The most widely used diagnostic tool for acute cerebral infarction is neuroimaging, which includes techniques such as computed tomography (CT) and magnetic resonance imaging. Diffusion weighted imaging has >90% sensitivity for diagnosing acute cerebral infarction, in particular [10]. In addition, currently many efforts are underway to enhance the diagnostic ability of neuroimaging tools; a study on biomarkers in acute cerebral infarctions is one example.
Biomarkers have shown efficacy in other medical fields, such as acute coronary artery syndrome, and can provide a great deal of information to assist with early diagnosis and intervention. This suggests that biomarkers can facilitate early therapeutic interventions when combined with neurological examination and neuroimaging in acute cerebral infarction [25,26].
Many studies have indicated that biomarkers linked to the ischemic cascade have a potential role in assessing acute cerebral infarction, such as early neurologic deterioration, final infarct volume, clinical status, and functional outcome [5,9]. However, to the best of our knowledge, no study has discovered a successful clinical biomarker. This may be because most proteins that increase during acute cerebral infarction do not have the major features that are required to be a significant and useful biomarker. These characteristics are : 1) brain specificity; 2) presence in plasma to enable blood, rather than cerebrospinal fluid, sampling; 3) presence in plasma samples at an early stage of onset of clinical manifestations; 4) expression that corresponds to the size of the lesion; and 5) prognostic value. Currently, the use of biomarkers for acute cerebral infarction has been confined to research laboratories owing to these constraints, and none has been used in a clinical setting [18].
To overcome these difficulties and enhance their diagnostic utility, studies have attempted to combine several biomarkers for use in a panel. Reynolds et al.19) and Lynch et al. [16] have shown that a panel of biomarkers can be cost-effective and time-saving in combination with brain imaging. This allows physicians to promptly identify subsets of patients who may benefit from immediate intervention and critical care.
There is debate regarding whether a panel of biomarkers can be clinically useful, have a good predictive rate, and aid pre-existing diagnostic tools [15]. Moreover, a limited number of biomarker studies have reported results during the early stages of acute cerebral infarction. This time-point is the critical window for treatment using intravenous recombinant tissue plasminogen activator (IV-tPA) within 4.5 hours, or interventional treatment options within 6 hours of stroke onset [3].
To create a panel of biomarkers that can be used during the acute stage of cerebral infarction, it is essential to discover and select biomarkers that can be detected in the plasma at the point of ischemic onset, and that are specific to ischemic brain injury. In this study, we selected S100β, matrix metalloproteinase-9 (MMP-9), D-dimer, and heat shock protein 70 (HSP70) from the proteins that are involved in various pathological processes during acute cerebral infarction, such as brain-specific markers, inflammatory mediators, thrombosis/hemostasis-related markers, and neuroprotective agents, to test their efficacy as clinically applicable biomarkers [15,16].
We used a mouse model of infarction with transient middle cerebral artery (MCA) occlusion to investigate the use of these proteins as biomarkers in acute cerebral infarction within 4.5 hours of symptom onset, the time when IV-tPA administration is feasible.
MATERIALS AND METHODS
Focal cerebral infarction in mice
Twenty-five C57BL/6 adult male mice (age, 7–8 weeks) weighing 23–25 g, were used in this study. The Institutional Animal Care and Use Committee reviewed and approved the animal protocols that were used herein. Mice were randomly assigned to three experimental groups : sham-operation (n=5), 30-minute middle cerebral artery occlusion (MCAO; n=10), and 60-minute MCAO (n=10).
Focal cerebral ischemia was induced by transiently occluding the MCA in these mice using a modified intraluminal filament technique [29]. Mice were anesthetized with 5% isoflurane in 30% O2/70% N2O using the V-10 Anesthesia system (VetEquip, Inc., Pleasanton, CA, USA). Following induction of anesthesia, the level of isoflurane was reduced and maintained at 1.5%. After a midline neck incision, the left common and external carotid arteries were isolated and ligated, and a microvascular clip was temporarily placed on the left internal carotid artery.
An 8–0 nylon monofilament coated with silicon resin was introduced through the incision into the common carotid artery and advanced 9 mm distal to the carotid bifurcation to achieve the MCAO. The nylon monofilament was removed after 30 or 60 minutes for mice in the 30- or 60-minute MCAO groups, respectively. Mice in the sham-operation group received a midline neck incision with left common and external carotid artery isolation and ligation, but did not undergo any further manipulation. The rectal temperature was maintained at 37±0.2℃ throughout the surgical procedure. After suturing the skin incision, the mice were returned to their cages.
Neurological scores
We evaluated the neurological deficits in each group 4 hours after the removal of the nylon filament. Mice from each group underwent behavioral assessment using a 6-point deficit score [11].
Scoring indications were as follows : 0, normal motor function; 1, mild circling behavior with or without inconsistent rotation when picked up by the tail and with <50% attempts to rotate to the contralateral side; 2, mild consistent circling with >50% attempts to rotate to the contralateral side; 3, consistent strong and immediate circling with the mouse maintaining a rotating position for more than 1–2 seconds, with its nose almost reaching its tail; 4, severe rotation progressing into barreling, loss of walking, or a righting reflex; and 5, a comatose or moribund state.
Measurement of cerebral infarction volume
Brains were extracted to identify the presence of infarction. Infarct volumes were measured in all mice. The brains were removed and sliced coronally into four 2-mm slices with a Brain Matrix (Braintree Scientific, Braintree, MA, USA) on ice. Slices were incubated in 2% 2, 3, 5-triphenyltetrazolium chloride and warmed to room temperature for 20 minutes, followed by fixation with 4% paraformaldehyde at 4℃ for 48 hours before scanning. The infarct area of each section was measured using National Institutes of Health (NIH) Image J software version 9 (National Institutes of Health, Bethesda, MD, USA), and the total infarct volume was calculated.
Ipsilateral and contralateral hemisphere volumes were also determined to correct for cerebral edema, which can artificially affect infarct volume [23]. The following formula was used to calculate the edema-corrected infarct volume : corrected infarct volume=contralateral non-infarct volume−ipsilateral non-infarct volume [24].
Assessment of plasma biomarkers (S100β, MMP-9, D-dimer, and HSP70) using enzyme-linked immunosorbent assay (ELISA)
For quantitative analyses of plasma S100β, MMP-9, D-dimer, and HSP70 expression, blood samples were obtained by cardiac puncture with a 1-mL syringe and 22-gauge needle 4 hours post-MCAO. Blood samples were collected in glass tubes with ethylenediaminetetraacetic acid, centrifuged at 1000 g for 30 minutes, and supernatants were frozen at -80℃ until further analysis. The amount of plasma necessary for each assay was calculated, and the optimal dilution ratios were determined (S100β, no dilution; MMP-9, 1 : 20; D-dimer, 1 : 4; HSP70, no dilution).
The plasma concentrations of S100β, MMP-9, D-dimer, and HSP70 were calculated using mouse protein S100β (CSB-EL-0206643MO; CUSABIO, Wuhan, China), mouse total MMP-9 (MMPT90; R&D Systems, Minneapolis, MN, USA), mouse D-dimer (E03D0224; BlueGene, Shanghai, China), and mouse HSP70 (CSB-E08311m; CUSABIO) ELISA kits. All sandwich ELISAs were performed using standard procedures according to the manufacturer’s instructions.
Sensitivity and specificity of the biomarkers
To evaluate the sensitivity and specificity of the biomarkers, we constructed a receiver operating characteristic (ROC) curve, which plotted test sensitivity as the y coordinate, and 1-specificity, or the false positive rate, as the x coordinate. We estimated the area under the ROC curve (AUC), which is measured between 0–1, since both axes have values ranging from 0–1. The best overall diagnostic performance was indicated by an AUC value of 1.
Statistical analysis
Statistical analyses were performed using the Statistical Package for the Social Sciences version 18.0 for Windows (SPSS Corp., Chicago, IL, USA). The Kruskal-Wallis and Mann-Whitney U tests were used to compare the distributions of continuous variables between groups. Two-sided hypothetical tests were used. The Spearman correlation coefficient was used to analyze the relationship between the biomarkers, infarct volumes, and neurological scores. A p value <0.05 was considered statistically significant.
RESULTS
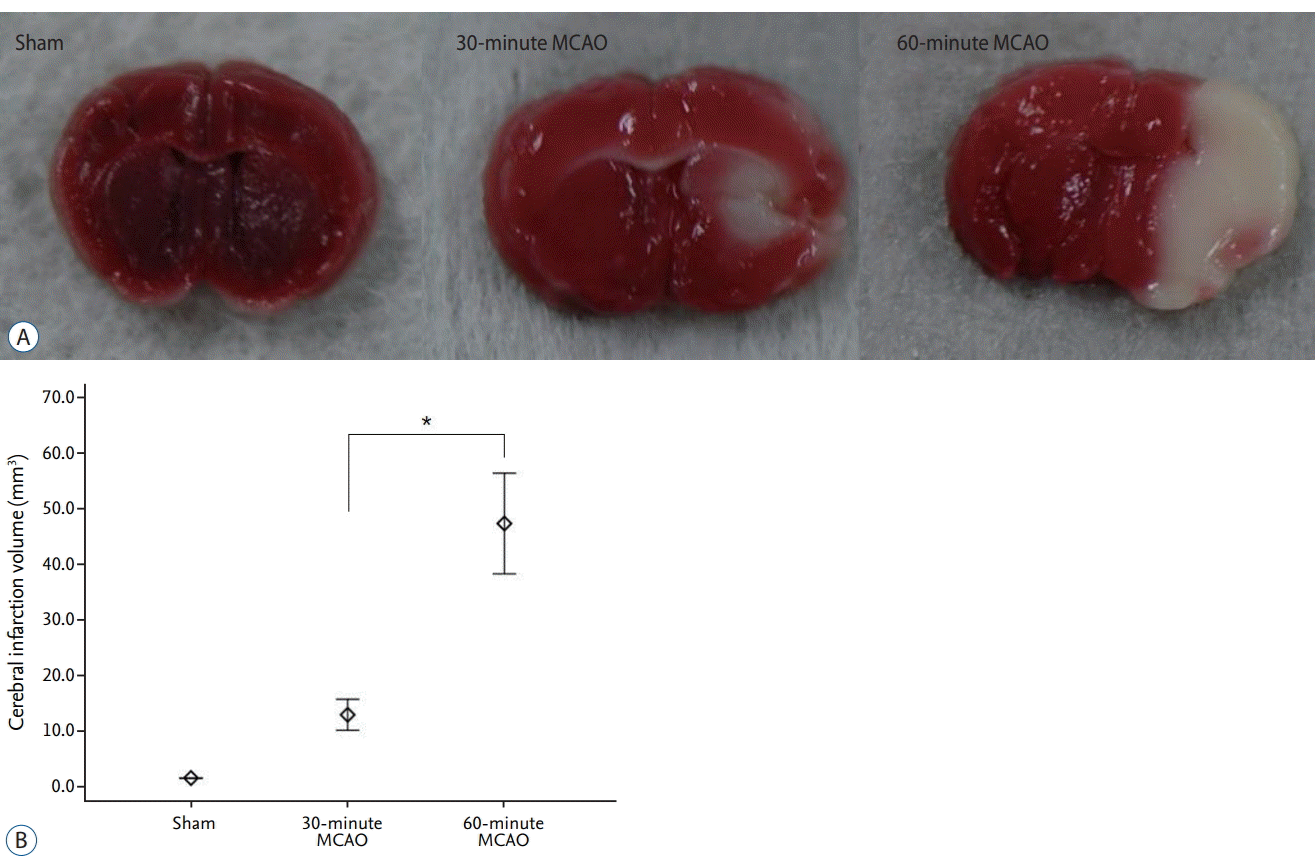
Cerebral infarction volume
Cerebral infarcts were found in the hemisphere ipsilateral to the occlusion in all mice that were exposed to transient ischemia (30-minute MCAO, n=10; 60-minute MCAO, n=10). No infarction was noted in mice from the sham group (n=5; Fig. 1A). The volume of cerebral infarction increased with MCA occlusion time. The average infarct volume was 12.32±2.31 mm3 in the 30-minute occlusion group, and infarcts were mainly confined to the basal ganglia. The average infarct volume was 46.9±7.43 mm3 in the 60-minute occlusion group, and the distribution was more extensive, affecting an area from the basal ganglia to the cerebral cortex. A significant difference in infarct volume was found between the 30-minute and 60-minute MCAO groups (p=0.008), with greater infarct volume in the 60-minute ischemia group (Fig. 1B).
Neurological score
Neurological deficits were found in all mice that underwent transient ischemia (30-minute MCAO, n=10; 60-minute MCAO, n=10). No neurological deficit was noted in mice from the sham group (n=5). The average neurological scores in the 30-minute and 60-minute ischemia groups were 1.6±0.55 and 3.2±0.70, respectively. The neurological score of the sham group was 0. A statistical comparison of the neurological scores revealed a signif icant difference between the 30-minute and 60-minute MCAO groups (p=0.002; Fig. 2).
Expression of plasma biomarkers
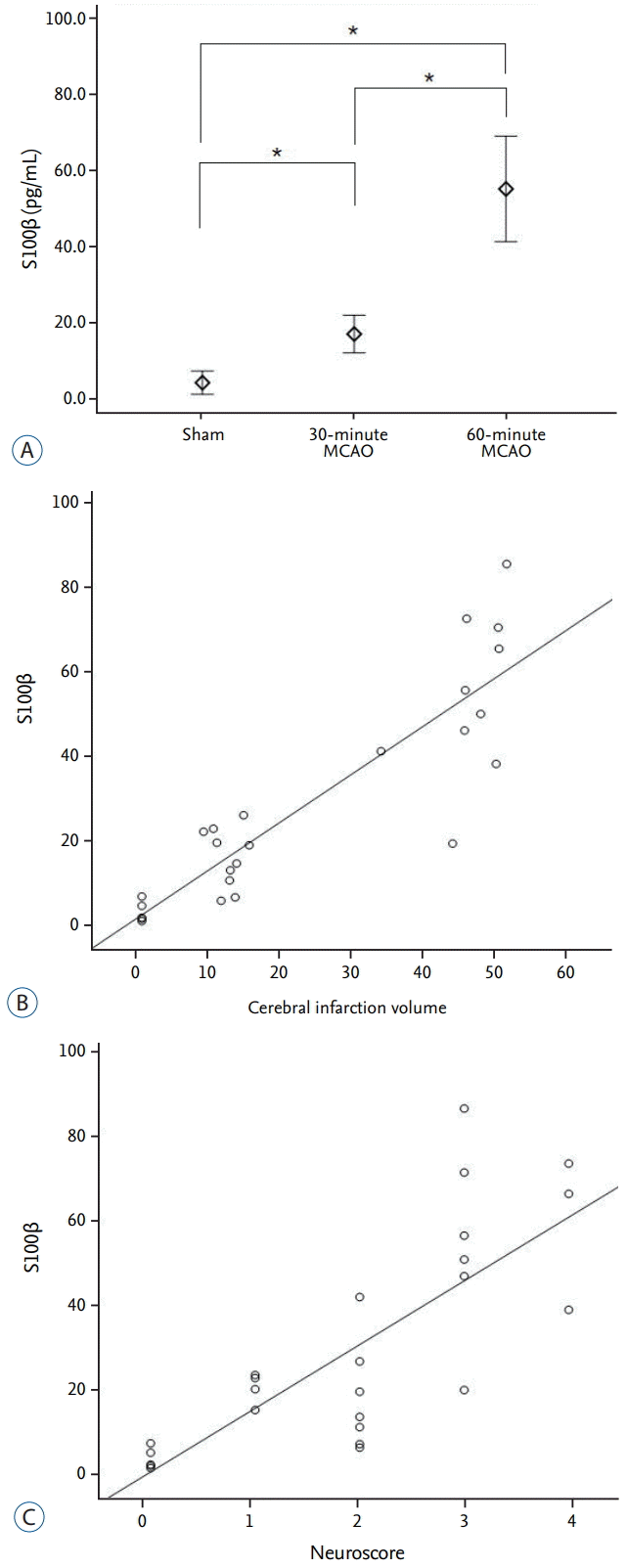
S100β
Quantitative ELISA measurements of plasma S100β showed average values of 3.9±2.47 pg·mL-1, 16.8±8.34 pg·mL-1, and 55.1±21.63 pg·mL-1 in the sham, 30-minute MCAO, and 60-minute MCAO groups, respectively. Thus, the concentration of S100β in both infarction groups was significantly increased when compared with the sham group (p=0.001). In addition, the 60-minute occlusion group had a significantly higher S100β concentration when compared with the 30-minute group (p=0.001; Fig. 3A). The plasma S100β concentration was significantly positively correlated with cerebral infarct volume and neurological score, with statistical values of r=0.867 (p<0.001) and r=0.802 (p<0.001), respectively (Fig. 3B and C).
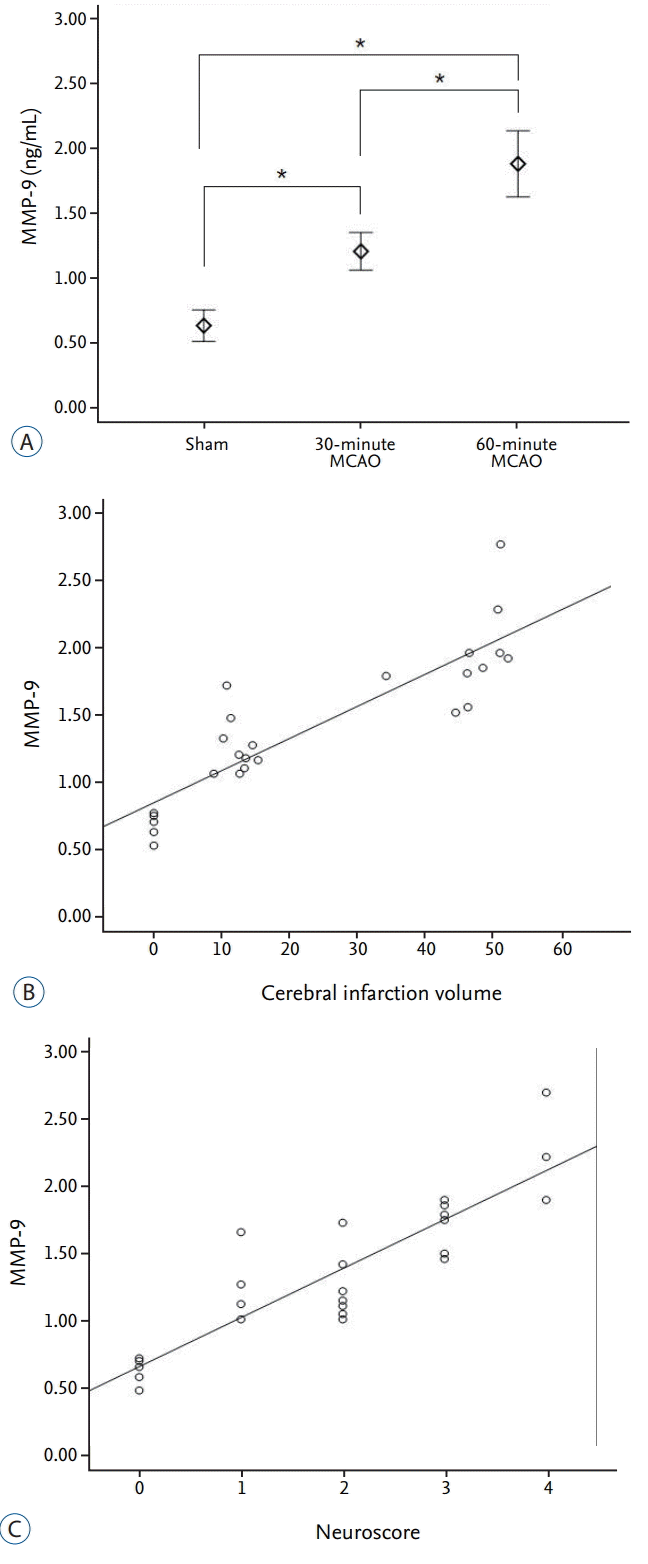
MMP-9
Quantitative ELISA measurements of plasma MMP-9 showed average values of 0.63±0.09 ng·mL-1, 1.20±0.22 ng·mL-1, and 1.88±0.41 ng·mL-1 in the sham, 30-minute MCAO, and 60-minute MCAO groups, respectively. MMP-9 concentration in both infarction groups was significantly increased when compared with the sham group (p=0.001). In addition, the 60-minute occlusion group showed a significantly higher MMP-9 concentration than the 30-minute occlusion group (p=0.001; Fig. 4A). The plasma MMP-9 concentration was significantly positively correlated with both cerebral infarct volume and neurological score, with statistical values of r=0.890 (p<0.001) and r=0.879 (p<0.001), respectively (Fig. 4B and C).
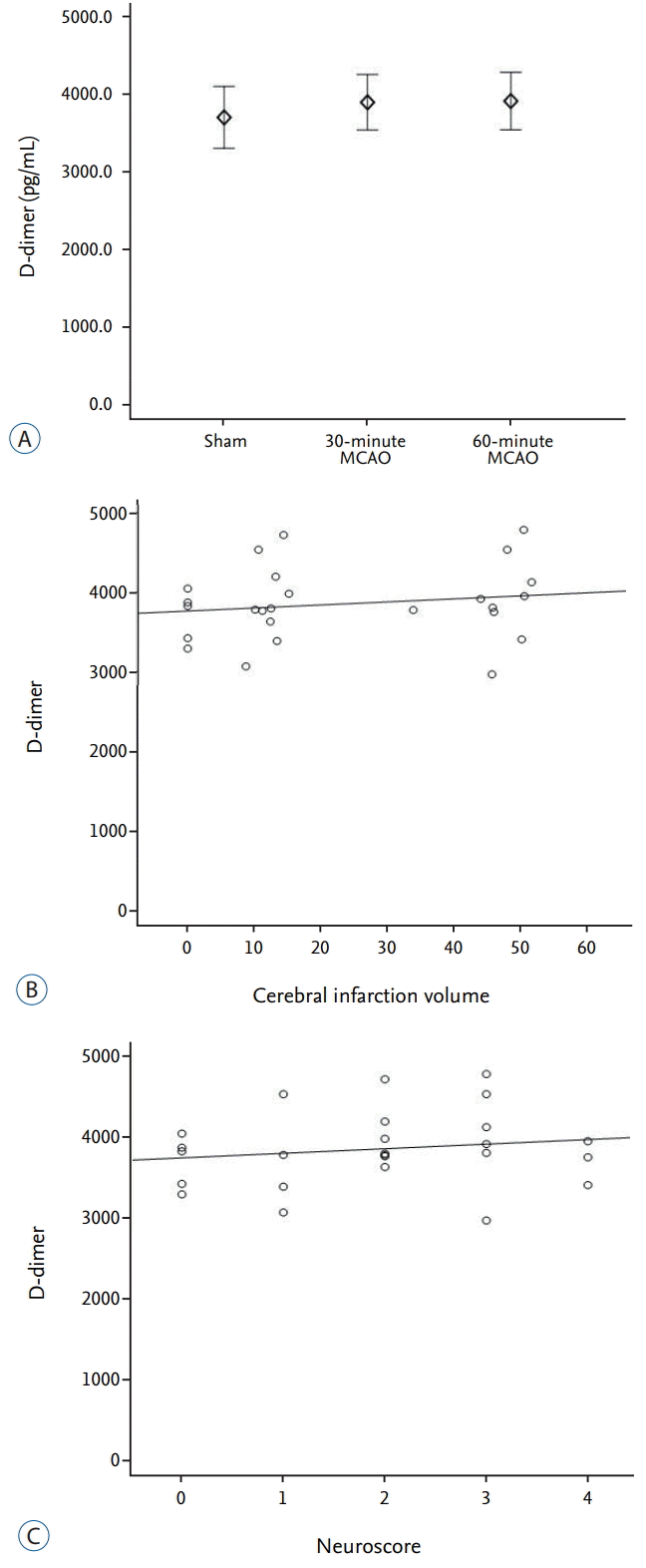
D-dimer
Quantitative ELISA measurements of plasma D-dimer showed average values of 3689±335 pg·mL-1, 3884±532 pg·mL-1, and 3900±1228 pg·mL-1 in the sham, 30-minute MCAO, and 60-minute MCAO groups, respectively. There was no significant difference in plasma D-dimer concentrations between the three groups (p=0.843; Fig. 5A). In addition, there was no significant correlation with cerebral infarct volume (r=0.255, p=0.218) or neurological score (r=0.139, p=0.507; Fig. 5B and C).
HSP70
Quantitative ELISA measurements of plasma HSP70 showed average values of 1.15±0.41 ng·mL-1, 2.48±0.15 ng·mL-1, and 2.24±0.19 ng·mL-1 in the sham, 30-minute MCAO, and 60-minute MCAO groups, respectively. The concentration of HSP70 in both infarction groups was significantly increased when compared with the sham group (p=0.001, for both). In addition, the HSP70 concentration was higher in the 30-minute occlusion group when compared with the 60-minute occlusion group, but this did not reach statistical significance (p=0.66; Fig. 6A). The plasma HSP70 concentration did not correlate with cerebral infarct volume (r=0.174, p=0.406) or neurological score (r=0.138, p=0.510; Fig. 6B and C).
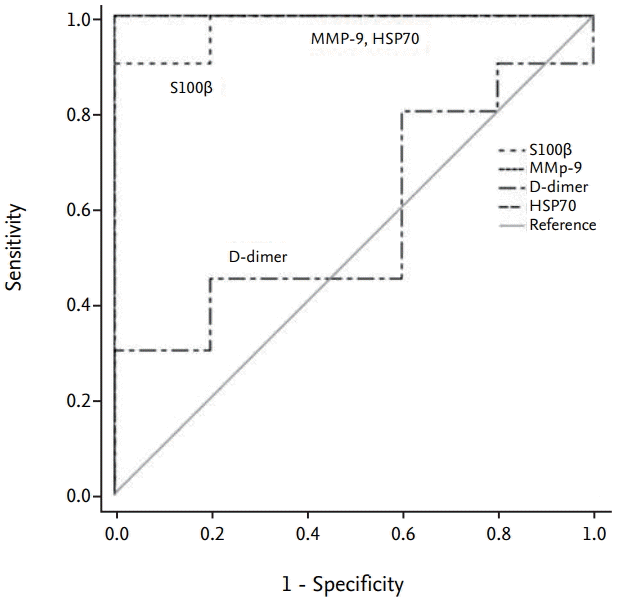
Sensitivity and specificity of the biomarkers
The area under the ROC curve (AUC) showed high sensitivity and specificity for MMP-9 and HSP70, which both showed an AUC of 1, and for S100β, which showed an AUC of 0.98. The AUC for D-dimer was 0.58 (Fig. 7).
DISCUSSION
The levels of many plasma proteins are elevated in patients with acute ischemic stroke [17]. These proteins are associated with certain biochemical changes that are involved in the pathogenesis of acute cerebral infarction.
The early phase of ischemia induces the activation of astroglial cells, neuronal damage, increased oxidative stress, and the release of inflammatory mediators. During the acute ischemic phase, brain-specific proteins are released into the bloodstream through blood-brain barrier (BBB) disruption, allowing for their serological detection. Additionally, the levels of inflammatory mediators and molecules related to thrombosis/hemostasis that are present in the bloodstream become elevated during the acute ischemic phase [25,26].
Despite theoretical evidence for these changes, no single biomarker has sufficient predictive value to serve as an early diagnostic indicator of ischemic stroke, or to distin guish stroke from other conditions. A recently published meta-analysis on stroke-related serological markers reported that each marker failed to improve the predictive value obtained in current clinical models [26]. Consequently, recent research has focused on enhancing diagnostic accuracy by establishing a panel of biomarkers to overcome the low sensitivity and specificity of individual biomarkers [15,26].
The Biomarker Rapid Assessment in Ischemic Injury Study group evaluated plasma D-dimer, S100β, MMP-9, and brain natriuretic peptide levels from patients within 24 hours of ischemic stroke or stoke-mimicking events. The group analyzed the diagnostic prediction rate and reported a diagnostic sensitivity of 90%. In addition, this rate was further increased when combined with CT [15]. To enhance the diagnostic accuracy and treatment efficacy of acute cerebral infarction, further research into an effective panel of biomarkers is required. To achieve this, highly sensitive and specific biomarkers for acute cerebral infarction need to be identified and selected.
S100β is a calcium-binding peptide secreted by astrocytes during brain injury, neurodegeneration, and psychiatric disorders. The interaction between S100β and the cerebral immune system leads to the enhanced survival of developing neurons at low concentrations of S100β. At higher concentrations, S100β stimulates the production of pro-inflammatory cytokines and apoptosis in vitro. Increased serum levels of S100β are found with ischemic stroke and many other acute and chronic conditions, such as traumatic brain injury, Alzheimer’ s disease, and schizophrenia [14].
In ischemic stroke, the serum concentration of S100β reaches its maximal concentration at days 2–4. Peak concentrations correlate with higher NIH Stroke Scale scores, infarct sizes, and the clinical course in both infarction and subarachnoid hemorrhage [6,7]. Reynolds et al. [19] reported that S100β concentration, which peaks within 0–12 hours of cerebral infarction, is highly relevant in ischemia. In addition, this pattern is found within 0–6 hours of stroke. Several other studies have reported a higher S100β concentration within 12 hours, or 6 hours at the earliest, of cerebral infarction onset when compared with that observed in control groups [6,15,19,27].
In our study, the concentration of S100β increased as ischemia developed, and further increased following a longer duration of MCAO. There was a positive correlation between S100β concentration and both infarct volume and the degree of neurological deficit. Overall, our results are consistent with previous reports that show that the concentration of plasma S100β can be used to complement traditional early diagnostic tools for acute cerebral infarction and to assess its severity.
MMPs are a family of 14 enzymes, including a subset that is active in brain tissue. MMPs are grouped into stromelysins, gelatinases, collagenases, membrane-type proteinases, and matrilysins [19]. MMPs are produced in latent form and require activation that leads to their toxicity. Once active, these enzymes are implicated in many different processes, such as disruption of the BBB, capillary damage, and prolonged inflammation. Consequently, they promote the secondary progression of ischemia and hemorrhagic transformation [4]. Both inflammation and alterations to the vascular wall structure due to MMP activity have been implicated in the chronic and progressive damage of intracranial atherosclerosis and acutephase injury [2].
MMP-9 is widely expressed in brain tissue. It can induce cerebral edema and hemorrhagic transformation via disruption of the BBB, thus increasing its permeability during ischemia. MMP-9 is produced by leukocytes, neurons, glial cells, vascular smooth muscle cells, and endothelial cells. It mediates inflammation by disassembling collagen IV, the basal lamina, and fibronectin. MMP-9 concentrations increase after 2 hours of ischemia, peak at 72 hours, and decrease 120 hours after onset. MMP-9 levels increase rapidly during cerebral infarction in accordance with the size of the infarct; therefore, it is a potential biomarker to aid with the early diagnosis and severity assessment of acute cerebral infarction [20].
This study suggests that the concentration of plasma MMP-9 can be used to complement current diagnostic protocols in the detection of early stage acute cerebral infarction and to estimate its severity. We found that the concentration of MMP-9 increased as ischemia developed, and higher concentrations were associated with a longer duration of MCAO. There was also a positive correlation with infarct volume and the degree of neurological deficit.
D-dimer, a marker of plasmin-mediated fibrin degradation, is cross-linked to fibrin degradation products (FDP) and indicates vessel occlusion. Plasmin splits fibrin into FDP and Ddimers when the coagulation and fibrinolytic system is activated. D-dimer is elevated under various conditions such as deep vein throm bosis, pulmonary thromboembolism, myocardial infarction, disseminated intravascular coagulation, surgery, trauma, and stroke [1].
An increase in the concentration of D-dimer during ischemic stroke is due to activation of the hemostatic system by meningeal irritation because of increased intracranial pressure, and activation of the coagulation pathway due to brain tissue injury and cerebrovascular disruption [8]. The plasma concentration of D-dimer increases 24 hours after cerebral infarction; however, we did not find any significant increase in the concentration of D-dimer following MCAO. In addition, it did not correlate with infarct volume or the degree of neurological deficit at the early stage of acute cerebral infarction.
HSP70 is a molecular chaperone induced by various pathological insults, such as hyperthermia, apoptotic stimuli, oxidative stress, and ischemia [29]. An increase in the gene expression of heat shock proteins, such as HSP70, in the brain is triggered by a variety of insults, including ischemia [18]. Yenari et al. [28] have suggested that increased HSP70 expression indicates cellular stress. Stressful conditions can result in the denaturing of proteins. The presence of these altered proteins triggers the transcription of heat shock proteins, such as HSP70, which bind to the denatured proteins to prevent further denaturation.
During cerebral infarction, HSP70 can be expressed in neurons, astrocytes, microglia, or endothelial cells in the brain [18]. Over-expression of HSP70 protects these cells against ischemic conditions by preventing protein denaturation, and stabilizing and restoring the functions of partially denatured proteins [18]. The HSP70 chaperone family regulates both apoptotic and necrotic cell death by blocking apoptosis at several different steps in the pathway. Based on this knowledge, we sought to verify whether HSP70 could serve as a biomarker of the acute stage of cerebral infarction. There was a significant increase in HSP70 following ischemia; however, this increase did not correlate with infarct volume or the degree of neurological deficit in our experimental setting.
As for this result, there is a clinical report implying that HSP 70 expression from lymphocytes correlated with both the severity of the stroke and improvement in neurologic deficit scores after stroke [12]. Therefore, animal and clinical studies will be need to investigate the relations of gene expressions from infarcted brain tissue and plasma during acute cerebral infarction.
In summary, S100β, MMP-9, and HSP70 were significantly increased during the acute stage of cerebral infarction induced by transient MCAO in our mouse model. In addition, the plasma concentration of S100β and MMP-9 correlated with the volume of cerebral infarction and the degree of neurological deficit. This suggests that they may be useful biomarkers in the diagnosis of acute cerebral infarction and the prediction of stroke severity.
One study has shown that D-dimer levels significantly increaseduring cerebral infarction and reflect stroke severity [22]. However, we did not find any meaningful increase in D-dimer concentration at the early stage of acute cerebral infarction. Further research is needed to clarify this.
HSP70 is expressed at low levels in the healthy brain but is induced in all cell types following ischemia, in particular in neurons in the penumbral region [21,23]. It has been demonstrated that HSP70 and other heat shock proteins are released by stressed cells as free soluble proteins or enclosed within detergent-soluble membrane vesicles [13]. Therefore, plasma HSP70 levels during the acute stage of cerebral infarction indicate ischemic stroke and predict the size of the penumbral region. Further studies including a large-scale prospective cohort study should be conducted to assess its clinical applicability as a biomarker.
There were several limitations in this study. First, the sample size was small and the number of biomarkers we assessed was limited. In addition, more categories of experimental infarction groups could have been used. Most mice did not survive to conduct a proper neurological examination when MCA occlusion was performed for longer than 1 hour; therefore, we excluded this group and stratified the mice into 30-minute and 60-minute MCAO groups and the evaluation of neurological deficits was based solely on a behavioral assessment. Second, we did quantitative analysis of protein expression with plasma during acute cerebral infarction in this study. To further enhance the diagnostic value, future study will be needed on comparative analysis of expression rates in the four groups of brain tissue and plasma, and also on the differences in gene expression according to infarction volume.
CONCLUSION
This study used a focal cerebral infarction model to verify S100β, MMP-9, and HSP70 as potential biomarkers in the diagnosis of cerebral infarction, estimation of infarction volume, and neurological deficit during the acute stage of the disease. If clinical research continues on to establish useful biomarkers expressed during acute cerebral infarction, there will come a new phase in the treatment of acute cerebral infarction, which will enable faster management of patients and more favorable clinical outcomes.
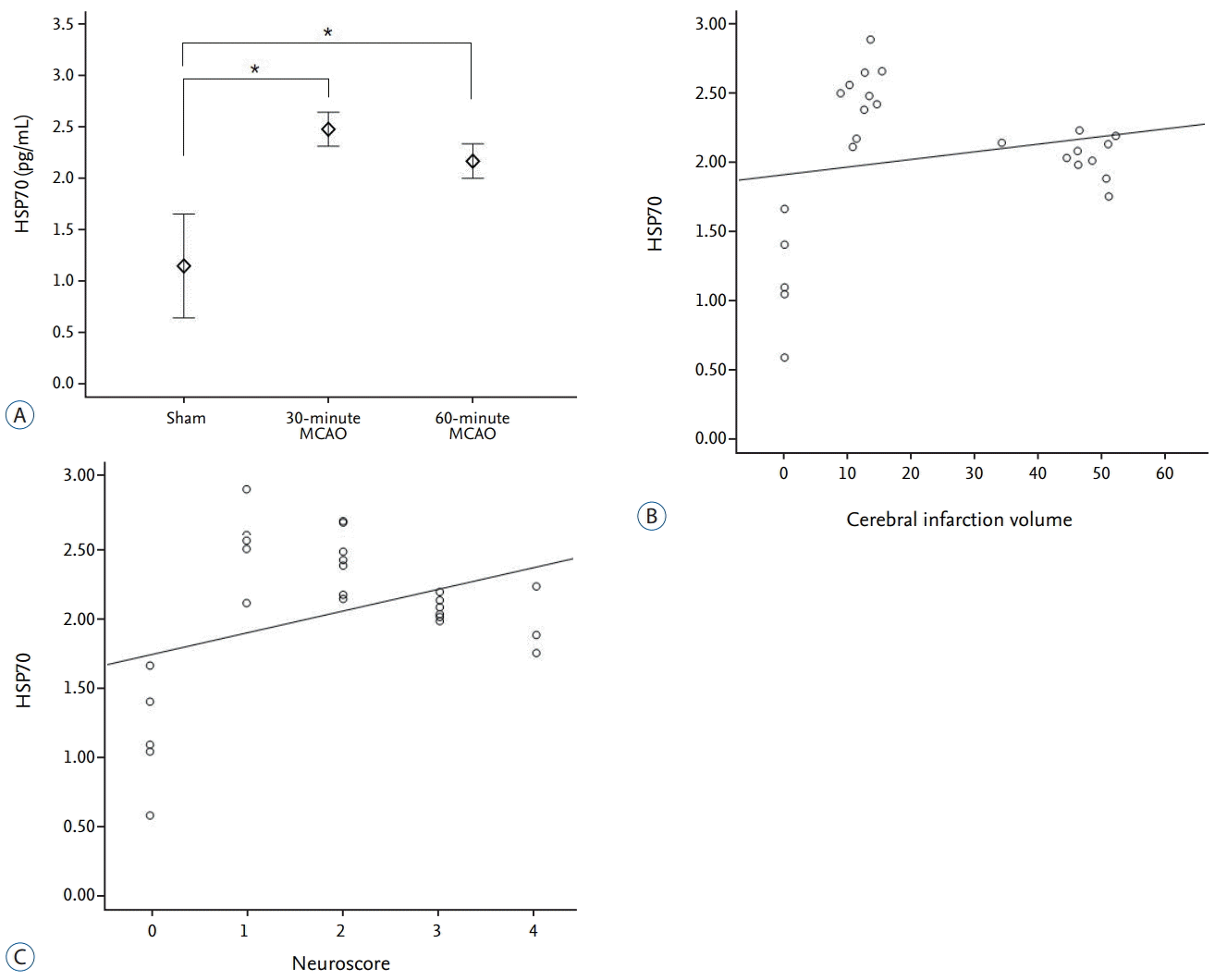
 XML Download
XML Download