

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Thyroid disorders and diabetes mellitus (DM) are common chronic endocrine disorders that often coexist. A higher prevalence of thyroid disorders is seen in patients with type 1 diabetes mellitus (T1DM) [1] and type 2 diabetes mellitus (T2DM) compared to the general population [2,3]. In addition, a higher prevalence of DM is noted in patients with thyroid disorders [4-6]. While increased medical surveillance in patients with DM or thyroid disorders may account for this trend, potential pathophysiological mechanisms do exist that might explain the development of thyroid disorders in patients with DM and also the increased risk for development of DM in individuals with thyroid disorders. Autoimmunity is an important element for understanding the linkage between T1DM and auto-immune thyroid disease (AITD) while the relationship between T2DM and thyroid disorders is more complex. The aim of this article is to review the links between alterations in glucose homeostasis, including diabetes, as related to the presence of thyroid dysfunction.
GLUCOSE HOMEOSTASIS AND THYROID HORMONE
Glucose homeostasis
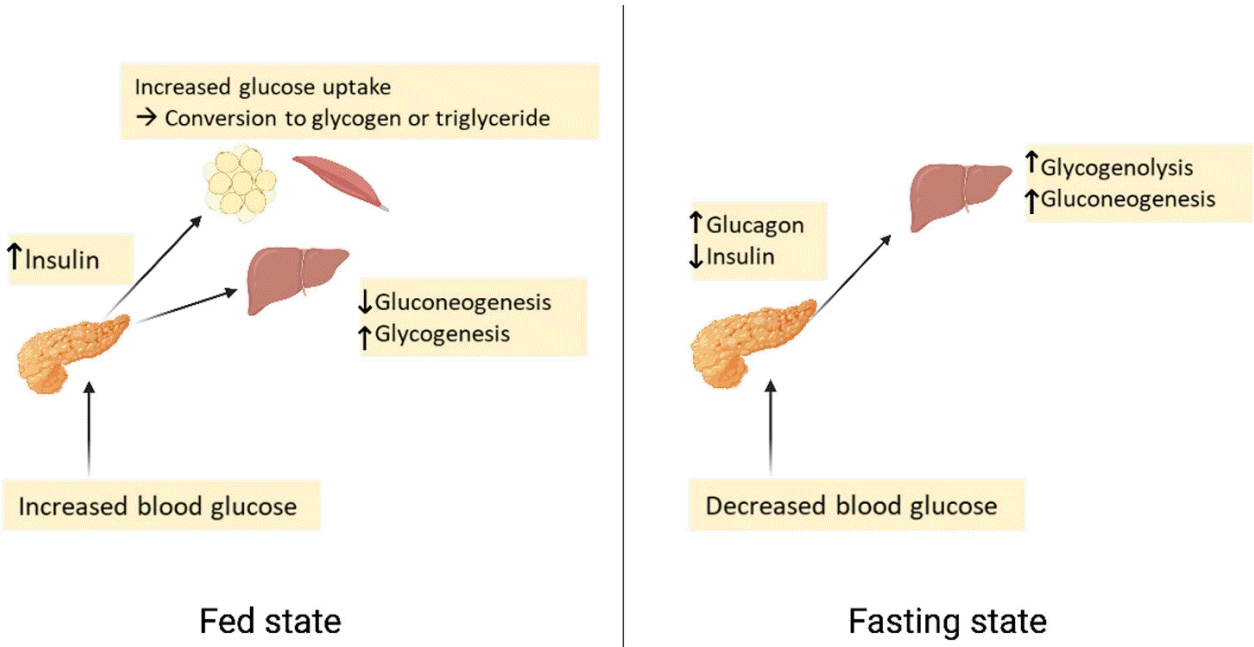
Plasma glucose levels are balanced by glucose entry and removal from the circulation. Glucose entering the circulation is derived from intestinal absorption during the fed state, glycogenolysis and gluconeogenesis. The rate of gastric emptying mainly determines the rate of glucose appearance in the circulation while hepatic breakdown of glycogen (glycogenolysis) and formation of glucose from non-carbohydrate substrates such as lactate, amino acids, and glycerol (gluconeogenesis) are other important sources of circulating glucose [7]. Glucose homeostasis is the tight regulation of blood glucose levels critical to maintain life in mammals. This is achieved by a balance of hormone and neuropeptides released from the brain, pancreas, liver, intestine, adipose, and muscle tissue [8]. Insulin and glucagon are the most important hormones for glucose homeostasis. Insulin is a pancreatic β-cell hormone that lowers blood glucose concentration in three ways: (1) promoting glucose uptake by the cells of insulin-sensitive peripheral tissues, including skeletal muscle and adipocytes; (2) promoting storage of glucose as glycogen, or conversion to fatty acids in the liver; and (3) suppression of postprandial glucagon secretion [7]. Glucagon is secreted from pancreatic α-cells and plays a major role in sustaining plasma glucose during the fasting state. When plasma glucose level falls below the normal level, glucagon secretion rises, leading to hepatic glucose production and return of plasma glucose to normal range [9]. Glucose homeostasis is also regulated by other glucoregulatory hormones like amylin, glucagon-like peptide-1 (GLP-1), gastric inhibitory polypeptide (GIP), epinephrine, cortisol, and growth hormone (GH). Amylin is isolated from pancreatic amyloid deposits and co-secreted with insulin by pancreatic β-cells in response to nutrient stimuli [10]. Amylin suppresses postprandial glucagon secretion, slows gastric emptying and reduces food intake and body weight [7]. GLP-1 and GIP, incretin hormones are secreted by intestinal L-cells. GIP stimulates insulin secretion but does not inhibit glucagon secretion or gastric emptying. GLP-1 not only stimulates glucose-dependent insulin secretion but also suppresses postprandial glucagon secretion. It also slows gastric emptying and reduces food intake and thereby body weight [11]. Blood glucose levels can also be sensed by the central nervous system [12]. In response to low plasma glucose, the ventromedial hypothalamus triggers release of counterregulatory hormones [13]. After a meal, glucose is absorbed and plasma glucose levels rise. Increasing glucose levels stimulate pancreatic β-cells to secrete insulin. Insulin increases glucose disposal by glycogen synthesis and lipogenesis in the liver, and promotes the uptake of glucose and conversion to glycogen or triglycerides by skeletal muscle and adipose tissue. In contrast, during a fasting state, glucagon is released to increase blood glucose levels through glycogenolysis. When fasting is prolonged, glucose is produced by hepatic gluconeogenesis (Fig. 1) [7].
The effects of thyroid hormone on glucose homeostasis
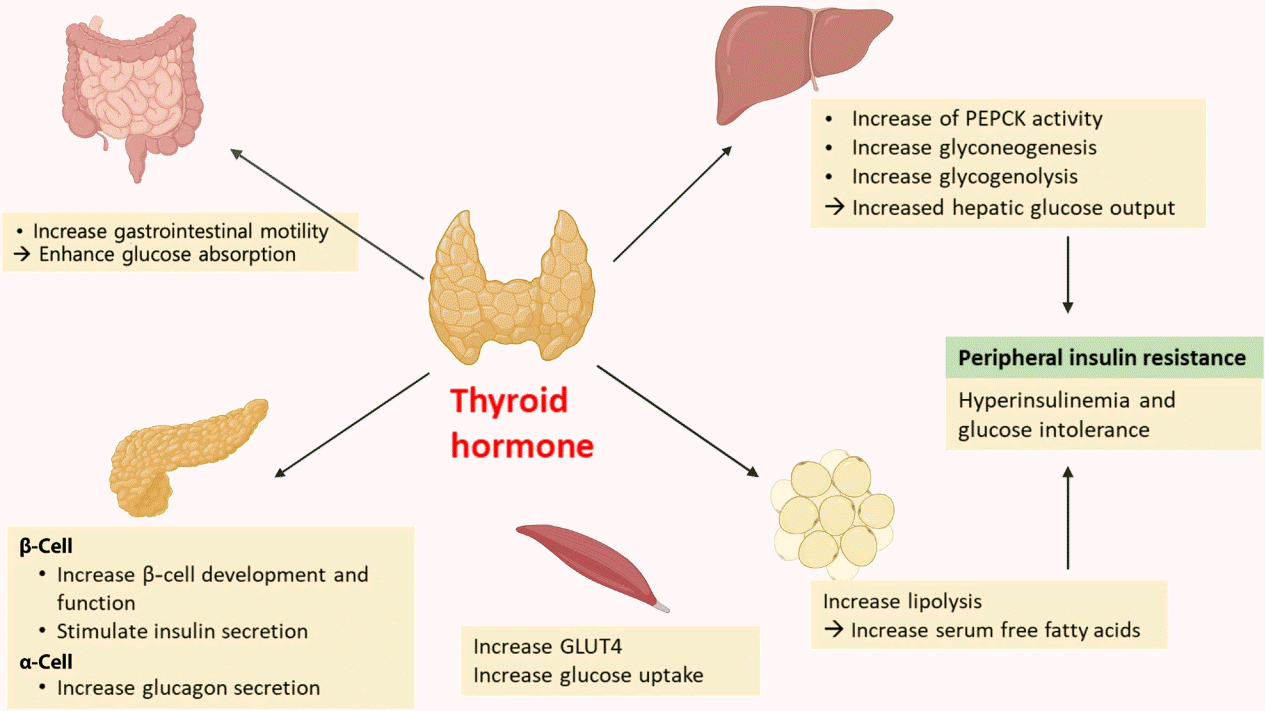
Thyroid hormone (TH) has long been known to affect glucose homeostasis. TH has been reported to be related to pancreatic β-cell development and influence glucose metabolism through several organs such as the liver, gastrointestinal tract, pancreas, adipose tissue, skeletal muscles, and the central nervous system (Fig. 2).
Thyroid hormone effects on the pancreatic β-cell
Several studies have shown links between TH action and pancreatic β-cell development and function [14,15]. Neonatal β-cells have thyroid hormone receptors (THRs). Aguayo-Mazzucato et al. [14] have described the role of TH and THRs in postnatal rat pancreatic β-cell development. Specifically, triiodothyronine (T3) stimulates β-cell proliferation from birth through the first week of life. THR also has a direct receptor-ligand interaction with the MAF bZIP transcription factor A (MAFA) promotor. In addition, T3 increases glucose-stimulated insulin secretion in vitro. In vivo, T3 administration acts on a pro-survival, anti-apoptotic factor for β-cells and improves glucose intolerance in streptozotocin-induced diabetic mice [15].
Peripheral effects of thyroid hormone on insulin secretion and resistance
TH impacts insulin secretion and glucose uptake via differing effects in the gastrointestinal tract, liver, skeletal muscles, and adipose tissue. TH enhances glucose absorption by increasing gastrointestinal motility [16]. In liver, it increases hepatic glucose output through increased hepatic expression of glucose transporter 2 (GLUT2) which stimulates the endogenous production of glucose through an increase in gluconeogenesis and glycogenolysis. T3 also increases hepatic gluconeogenesis by increasing activity of phosphoenolpyruvate carboxykinase (PEPCK), an enzyme that enhances gluconeogenesis [17,18]. Furthermore, stimulated glycogenolysis and gluconeogenesis induces hyperinsulinemia and glucose intolerance, resulting in peripheral insulin resistance [16]. In adipose tissue, TH increases lipolysis, resulting in an increase in free fatty acid that stimulates hepatic gluconeogenesis [19]. TH also directly stimulates insulin secretion by pancreatic β-cells and increases glucagon release by pancreatic α-cells [16]. Hyperthyroidism increases glucose transporter type 4 (GLUT4) gene expression and glucose uptake in skeletal muscles [16].
Central interactions of thyroid hormones on glucose and lipid regulation
T3 can centrally modulate glucose production through a sympathetic pathway from the hypothalamic paraventricular nucleus (PVN) to liver. T3 in the hypothalamic PVN increases hepatic glucose production, independent of glucoregulatory hormones [20].
THYROID DISORDERS AND DIABETES MELLITUS
Thyroid disorders and DM are closely linked. Many studies have demonstrated the higher prevalence of thyroid dysfunction in both T1DM and T2DM. The prevalence of thyroid disorders in a patient with DM is variable among the population studied. A large clinical study of 1,310 adult patients with diabetes reported a 13.4% overall prevalence of thyroid diseases. The prevalence was 31.4% in T1DM and 6.9% in T2DM, and 8.8% in men and 16.8% in women [21]. A meta-analysis of all available data in 10,920 patients with DM revealed an 11% prevalence of thyroid diseases. In this study, there was no difference between T1DM and T2DM, but the prevalence in women was more than twofold than that in men [22]. The fact that thyroid disorders increase the risk of DM has been reported in nationwide Danish health register study as follows. In an observational cohort study of 2,631 hyperthyroid singletons and 375 twin pairs discordant for hyperthyroidism, hyperthyroid individuals were 43% more likely to be diagnosed with DM [4]. In separate observational cohort study of 2,822 individuals with hypothyroidism, the group with hypothyroidism had a 40% higher prevalence of DM compared to controls [6].
Thyroid disorders and T1DM
Association of AITD and T1DM
T1DM accounts for 5% to 10% of diabetes cases and stems from cell-mediated autoimmune destruction of pancreatic β-cells [23]. Related autoimmune markers include autoantibodies to insulin, glutamic acid decarboxylase 65 (GAD65), the tyrosine phosphatase-related islet antigen-2 (IA-2) and IA2b, and zinc transporter 8 (ZnT8). Patients with T1DM are also at increased risk for development of other autoimmune disorders, such as: Hashimoto thyroiditis, Graves’ disease, celiac disease, Addison disease, autoimmune hepatitis, vitiligo, myasthenia gravis, and pernicious anemia [1]. AITD and T1DM are related target-organ autoimmune diseases with AITD being noted in 17% to 30% of adults with T1DM and with increased risk being observed in both autoimmune hypothyroidism and hyperthyroidism [24]. This association is linked to the expression of thyroid autoantibodies like thyroid peroxidase antibody (TPO Ab) and thyroglobulin antibody (TG Ab) [25,26]. A study that evaluated the prevalence of various autoantibodies in a population of 814 individuals with T1DM revealed that TPO Ab and Tg Ab were the most common autoantibodies with a proportion of 29% [27].
There is significant evidence for a genetic association between the two diseases [28]. These two diseases frequently occur within the same family and even within the same individual. AITD and T1DM often occur in the same individual or more than one member of the same family, as the phenotype classified as one of the autoimmune polyglandular syndrome 3 variant (APS3), especially APS type 3A [29,30]. Both T1DM and AITD are multifactorial autoimmune endocrine diseases with several susceptibility genes and environmental factors contributing to disease etiology. The human leukocyte antigen (HLA) class II, cytotoxic T lymphocyte antigen-4 (CTLA-4) (on chromosome 2q33), protein tyrosine phosphatase non-receptor type 22 (PTPN22) (1p13), forkhead box P3 (FOXP3) (Xp 11), and interleukin 2 receptor α (IL-2Rα) (10p15) are major genes found to contribute to a joint susceptibility for T1DM and AITD [29,31,32]. These genes are immunologically linked, influencing T cell activity. The HLA-DR molecules present autoantigens to T cells, and CTLA-4 is expressed on T cells and acts as a costimulatory receptor which downregulates T cells. PTPN22 is a negative regulator of T cell receptor signaling pathway, and FOXP3 regulates the differentiation of regulatory T cells, while IL-2Rα actively suppresses autoreactive T cells via CD25 (ɑ chain of the high-affinity IL-2 receptor) [29,32].
Screening recommendation for thyroid dysfunctions in patients with T1DM
Thyroid dysfunction is especially common in T1DM, particularly those with positive TPO Abs [25]. In these patients, thyroid diseases may be asymptomatic or the symptoms may be masked by symptoms related to poor diabetic control. Thus, a symptom-based approach to thyroid disease can miss the diagnosis in T1DM. Routine screening may help detect thyroid disease in such scenarios. Currently, screening for thyroid dysfunction is recommended in children, adolescents, and adults with T1DM by the American Thyroid Association (ATA) [33], the British Thyroid Association (BTA) [34], the American Diabetes Association (ADA) [35], the American Association of Clinical Endocrinologist (AACE) [36], National Institute for Health and Care Excellence (NICE) [37], and the International Society for Pediatric and Adolescent Diabetes (ISPAD) [38] but not by the United States Preventive Service Task Force (USPSTF) (Table 1) [39].
Thyroid disorders and T2DM
Thyroid disorders and insulin resistance
The glucose homeostasis disorder, insulin resistance, is defined as the decreased metabolic response to insulin in the peripheral tissues such as muscle, liver, and adipose [40]. To maintain glucose homeostasis, pancreatic β-cells compensate for insulin resistance by augmenting insulin secretion, leading to a state of chronic hyperinsulinemia. Insulin resistance plays a role in the pathogenesis of T2DM. Thyroid function is associated with insulin resistance not only in clinically diagnosed DM but also in subjects with a normal glucose tolerance [41,42]. Both hyperthyroidism and hypothyroidism can affect insulin resistance, although by different mechanisms.
Thyrotoxicosis and T2DM
The prevalence of hyperthyroidism is higher in patients with diabetes than in the general population. Approximately 4.4% of adults with T2DM also had hyperthyroidism compared to an average of 1.3% in the general United States population [2,21]. Thyrotoxicosis has been recognized to lead to hyperglycemia via several mechanism [43]. Excess TH can increase glucose absorption by the gastrointestinal tract, glucose production in the liver via increasing gluconeogenesis and glycogenolysis, and the concentration of free fatty acids via promoting lipolysis [16]. TH stimulates insulin secretion and induces hyperinsulinemia, although thyrotoxicosis also accelerates insulin degradation and decreases the half-life of insulin [44]. Furthermore, the increased hepatic glucose output induces hyperinsulinemia, glucose intolerance, and peripheral insulin resistance. Thus, hyperthyroidism precipitates impaired fasting glucose and/or diabetes and worsens glycemic control in pre-existing T2DM. Moreover, thyrotoxicosis may rarely precipitate diabetic ketoacidosis not only in patients with T1DM, but also in patients with T2DM when accompanied by insulin deficiency which can enhance the risk for lipolysis [16,45,46].
Hypothyroidism and T2DM
The prevalence of hypothyroidism is higher in patients with diabetes than in the general population. The prevalence of hypothyroidism in T2DM has been reported to be between 5.7% and 25.3% [47-49]. These wide range could be explained by differences in age, sex, and the degree of iodine intake of the populations surveyed [50]. The prevalence of hypothyroidism increased in older age, female gender, with family history of thyroid disease and positive TPO Ab [51].
Hypothyroidism is associated with insulin resistance and glucose intolerance with treatment of hypothyroidism and a return to a euthyroid states being associated with improved insulin sensitivity [41,52,53]. The pathogenesis of insulin resistance in a hypothyroid state is different from that seen with thyrotoxicosis. Hypothyroidism results in impaired glucose absorption from the gastrointestinal tract, delayed peripheral glucose assimilation, and gluconeogenesis [41]. Insulin resistance is thought to be associated with decreased glucose disposal, a result of decreased skeletal muscle and adipose tissue sensitivity to insulin [54,55]. This deterioration of glucose metabolism is also seen in subclinical hypothyroidism. One meta-analysis study revealed that the risk of developing subclinical hypothyroidism increased by 1.93-fold in T2DM patients compared to non-diabetics and that subclinical hypothyroidism may also be associated with an increase in diabetic complications [56]. Free thyroxine (T4) levels in the lower end of reference range are also associated with higher glycosylated hemoglobin (HbA1c) levels in euthyroid state [57].
Screening recommendation for thyroid dysfunction in patients with T2DM
Contrary to the guideline for T1DM, there is a lack of recommendations for screening for thyroid disease in patients with T2DM. The ATA recommended that adults who are at least 35 years old should be screened for thyroid disorders by measuring a thyroid-stimulating hormone (TSH) concentration and then every 5 years in the 2010 guidelines. Although there is no mention of DM type, the guideline recommends more frequent tests in those with high risk factors like diabetes [33]. The United Kingdom 2006 guidelines recommended a thyroid function test at the time of diagnosis for patients with T2DM but not annual testing [34]. Conversely, USPSTF reports that there is insufficient evidence to recommend thyroid dysfunction screening in non-pregnant or asymptomatic adults in 2015 [39]. There is no mention of monitoring thyroid function in T2DM in the 2015 NICE guidelines [58], the ADA guideline [59], the ATA/AACE guideline [60], and the ISPAD guideline (Table 2) [61,62]. Therefore, the necessity of a screening for thyroid dysfunction in patients with T2DM is debated at this time.
Thyroid disorders in pregnant women with diabetes
Changes in thyroid function during pregnancy and effect on glucose homeostasis
During pregnancy, numerous hormonal and metabolic changes occur, which affect glucose metabolism and thyroid function [63]. In a normal pregnancy, a 50% to 60% decrease in insulin sensitivity occurs in both pregnant women with normal glucose tolerance and women with gestational diabetes. In women with normal glucose tolerance, the decrease of insulin sensitivity is overcome by increased insulin secretion from pancreatic β-cells, whereas gestational diabetes occurs when insulin secretion is not sufficient to match this higher demand [64].
During pregnancy, the maternal thyroid gland increases in size by 10% in iodine replete countries but by 20% to 40% in iodine deficient countries [65]. Production of THs, T4, and T3 increases by almost 50% and for this reason, iodine daily intake should also be increased by 50%. The World Health Organization recommends daily iodine intake of 250 μg for pregnant and lactating women [66]. In early pregnancy, the placenta makes human chorionic gonadotropin (hCG) which is a member of the glycoprotein hormone family that is composed of common α-subunits and a hormone-specific β-subunits. The hCG α-subunit sequence is identical to the sequences of the α-subunits of TSH, follicle-stimulating hormone and luteinizing hormone [67]. Increased hCG concentration during the first trimester of pregnancy stimulates the thyroid gland thereby increasing production and release of THs resulting in decrease of TSH [66]. Hyperemesis gravidarum is extreme, persistent nausea and vomiting during pregnancy which can lead to dehydration, weight loss of at least 5% from pre-pregnancy baseline, ketonuria, and electrolyte imbalances. The exact cause is not fully understood, but it is believed to be caused by a rapid rising blood level of hCG [68,69]. hCG is a thyroid stimulator and most transient non-immune hyperthyroidism in early pregnancy is related to increased hCG levels while rare cases of familial recurrent gestational hyperthyroidism are caused by a mutant thyrotropin receptor that is hypersensitive to hGC as has been reported with even normal range serum hCG [70].
Thyroid dysfunction and DM are the common endocrinopathies during pregnancy and can affect both maternal and fetal health [71,72]. Thyroid function abnormalities are more common in pregnant women in the first trimester [73] with hypothyroidism, hyperthyroidism, and thyroid autoimmunity being common conditions in pregnancy, with prevalence rates of 2%–3%, 0.1%–0.4%, and up to 17% [74-76]. A higher prevalence of hypothyroidism in women with gestational diabetes mellitus (GDM) has been reported [77] and this association is also seen in postpartum thyroiditis (PPT), which is an autoimmune-mediated destructive thyroiditis in the 1st year postpartum. According to the studies, PPT is also three to four times more common in women with T1DM than in healthy women [78-80].
Considering that THs affect glucose homeostasis, thyroid disorders have been thought to affect the development of gestational diabetes or glucose control of pregnant women with diabetes. Pregnant women with either overt hypothyroidism or subclinical hypothyroidism have an increased risk of GDM [77,81]. There are also reports that maternal isolated hypothyroxinaemia (normal TSH concentration in conjunction with a low free T4) is associated with adverse metabolic parameters including increased maternal body mass index, higher fasting and postprandial glucose, higher HbA1c, higher triglycerides and increased insulin resistance (homeostasis model assessment of insulin resistance) in pregnancy [82,83]. Recently, there is a report that higher free T3 levels in early pregnancy may correlate with a greater risk for GDM developing compared to women who have normal levels of this hormone [84].
Screening recommendation for thyroid dysfunction in pregnant patients
Untreated overt hypothyroidism and hyperthyroidism are associated with adverse obstetrical and fetal outcomes such as spontaneous abortion, preeclampsia, preterm birth, abruptio placentae, poor fetal growth, and stillbirth [85,86]. Subclinical hypothyroidism has been related to adverse obstetrical outcomes such as abruptio placentae, preterm birth, and impaired neurodevelopment in offspring [87,88]. However, detection and treatment of maternal subclinical hypothyroidism has also not been shown to cause improved pregnancy outcome and neurocognitive function in offspring [89,90]. Therefore, clinical guidelines do not recommend universal thyroid function screening in pregnant women, but instead limited to women who have signs and symptoms of possible thyroid dysfunction and high-risk groups including those with T1DM or family history of thyroid disease. Similarly, screening for thyroid dysfunction is recommended in pregnant women with T1DM by the ATA [66], the BTA [34], the Endocrine Society [91], the AACE [60], the Korean Thyroid Association (KTA) [92], and the American College of Obstetricians and Gynecologists (ACOG) (Table 3) [93].
ANTIDIABETIC AGENTS AND THYROID DISORDERS
The use of certain antidiabetic drugs in patients with T2DM may influence thyroid function. We explore common oral antidiabetic agents and their impact on TH levels here (Table 4).
Metformin
Metformin is a wildly used oral antidiabetic drug for treatment of T2DM. Metformin suppresses hepatic gluconeogenesis via stimulating adenosine monophosphate-activated kinase (AMPK) in the liver, but has the opposite effect inhibiting AMPK in the hypothalamus [94]. Metformin has been shown to cross the blood-brain barrier and its concentrations in the hypothalamus match those in plasma in rat experiments, and substantially metformin levels in the pituitary gland increase as well [95]. These effects may fit with some clinical results, indicating that metformin may inhibit pituitary TSH secretion. It was first reported that metformin therapy decreases serum TSH levels in patients with T2DM with primary hypothyroidism in 2006 [96]. Several studies have shown that metformin administration in diabetic patients with primary hypothyroidism is associated with a reduction in serum TSH levels and this effect was observed in both patients under TH replacement therapy and in untreated patients [97,98]. The TSH-lowering effect of metformin was observed in patients with overt hypothyroidism and subclinical hypothyroidism, but not in euthyroid patients [98,99]. Despite the reduction in TSH, there is no change in [96] and plasma free T4 and free T3 concentrations, and the effect was not associated with clinical hyperthyroidism. In addition, this effect was reversible and usually resolved by 3 months after discontinuation [97,98].
Metformin has been shown to have beneficial effects in various types of cancers such as colon, rectum, pancreas, breast and prostate cancer [100]. Metformin inhibits the mammalian target of rapamycin (mTOR) activity by activating liver kinase B1 (LKB1) and AMPK, and thus prevents protein synthesis and cell growth. Metformin also can activate p53 by activating AMPK and thereby ultimate stop the cell cycle [101,102]. One study showed that metformin significantly decreased thyroid nodule size by 30%–50% in patients with insulin resistance [103]. Studies on the anti-tumor effect of metformin in thyroid cancer have been conducted, and some studies have reported that metformin inhibits thyroid cancer cell growth and induces cell-cycle arrest and apoptosis through AMPK activation and mTOR inhibition in vivo and in vitro [104-107]. A retrospective cohort study of 128,453 Korean adult diabetic patients reported that thyroid cancer decreased significantly in metformin users compared to metformin nonusers [108]. These findings suggest that metformin could be of potential benefit in thyroid cancer, especially in diabetic patients, but additional studies are required.
Sulfonylureas
Sulfonylurea drugs have been used in the treatment of T2DM since 1950’s. These drugs have been reported to have both antithyroid and goitrogenic activity. Early studies showed that large doses of sulfonylureas increase the weight of the thyroid gland and reduced iodine content and fixation of radioiodine uptake in animal models [109,110]. It has been reported that hypothyroidism occurs more frequently in diabetic patients taking first-generation sulfonylureas than in patients treated with diet alone or insulin [111]. However, studies on the second-generation sulfonylureas, glibenclamide, and gliclazide have shown that the use of these agents appear to have no influence on TH metabolism [112,113].
Thiazolidinediones
Thiazolidinediones (TZDs) are an important class of insulin sensitizers used in the treatment of T2DM. TZDs were known to act by increasing the transactivation activity of peroxisome proliferator-activated receptors (PPARs). TZDs are agonists of PPAR-γ, can reduce hepatic glucose production, increase peripheral glucose utilization and lipid metabolism [114]. PPAR-γ is found predominantly in adipose tissue and plays a dominant role in adipocyte differentiation [115]. PPAR-γ is also strongly expressed in thyroid tissue of patients with thyroiditis and Graves’ disease and in the orbital tissue of patients with thyroid eye disease (TED) [116]. TED is characterized by swelling of extraocular muscles and an extension of adipose tissue volume within the confines of the bony orbit [117]. TSH receptor expression in TED orbital preadipocyte fibroblasts is linked to adipogenesis, and stimulation of the PPAR-γ receptor results in differentiation of these cells. Rosiglitazone treatment of orbital preadipocyte fibroblasts from patients with TED revealed stimulation of both adipocyte differentiation and TSH-dependent cyclic adenosine monophosphage production in these cells [117]. In some patients with T2DM, it has been reported that eye protrusion can worsen while taking TZDs [118,119]. Thus, TZDs should be used carefully in patients with T2DM with clinically active TED.
The result from one study suggests that rosiglitazone administration in T2DM may reduce the risk of thyroid cancer [120]. The relationship between TZDs and thyroid cancer, however, has not been clearly elucidated.
Incretin mimetics: GLP-1 receptor agonists and dipeptidyl peptidase-4 inhibitors
Incretin mimetics are agents that act like or increase endogenous incretin hormones, including glucagon-like peptide-1 receptor (GLP-1R) agonists and dipeptidyl peptidase 4 (DPP4) inhibitors, respectively. GLP-1R agonists have shown association with C-cell proliferation and potential for increased risk of medullary thyroid cancer (MTC) in preclinical studies in rats [121-123]. Long-term dosing with liraglutide causes C-cell hyperplasia, calcitonin production in a dose dependent manner, and tumor formation in rodents [121-123]. This effect, however, was not seen in monkeys and humans. Prolonged use of liraglutide at very high doses did not produce C-cell proliferation in monkeys and did not induce significant calcitonin level changes in human clinical studies [124,125]. Exenatide also did not increase calcitonin in follow-up [126]. Similarly, in the Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes (LEADER) Trial, liraglutide did not increase calcitonin or cause C-cell malignancies [127]. This may be due to C-cells in monkeys and humans having lower expression of GLP-1R and decreased responsiveness to GLP-1R agonists compared to rodents [125]. The Food and Drug Administration (FDA) does not recommend monitoring for the occurrence of MTC in patients taking GLP-1R agonists; however, the use of GLP-1 R agonists in patients with a personal of family history of MTC or multiple endocrine neoplasia (MEN) type 2 is not recommended.
The relationship between DPP4 inhibitors and thyroid cancer has not been well reported. One study with sitagliptin among Taiwanese T2DM patients showed an increased prevalence of thyroid cancer. This was during the first year of treatment; however, and the impact of DPP4 inhibitor to this finding is thought to not necessarily be directly related given this short time interval and lack of additional supportive data [128].
Incretin mimetics have been shown to alter TSH concentration; however, this does not appear to be clinically significant. The GLP-1R agonist, exenatide, decreased TSH concentrations in diabetics after 6 months. Levels remained in the detectable range, and there were no significant changes in free T4, free T3, or thyroid volume [126]. A small study reported that diabetics followed for over three years taking a DPP4 inhibitor compared to diabetics on alternative therapy had higher TSH concentrations. TSH was still within the normal range and free T4 concentration was not significantly different between the two groups [129]. Larger, longer-term studies are warranted.
Colesevelam
Bile acid sequestrants (BASs) were developed as lipid-lowering agents for the treatment of hypercholesterolemia and include cholestyramine, colesevelam, colestilan, colestimide, and colestipol. BASs significantly reduce low-density lipoprotein cholesterol levels and improve cardiovascular outcomes in a population of asymptomatic middle-aged men with primary hypercholesterolemia [130]. BASs also lower glucose levels in patients with T2DM [131-134]. Among BAS, colesevelam was approved in 2008 by the U.S. FDA to improve glycemic control in adults T2DM patients [135]. TH including T4 and T3 are secreted into the bile. BASs can sequester T4 in the intestine and increase its fecal excretion, thus, restricting the enterohepatic reabsorption into the systemic circulation. Cholesytramine for example, is used for the treatment of thyrotoxicosis including thyroid storm because of this effect [136]. BSA agents can enhance the clearance of THs and may reduce the blood levels and effects of levothyroxine by interfering with the absorption of the drug. Cholestyramine and colestipol, inhibit absorption of levothyroxine through the binding of T4 to the basic anion copolymer resins in the gastrointestinal tract. Colesevelam is a nonabsorbed polymer that binds to bile acids in the small intestine, thereby preventing their reabsorption. Although it has been proposed that the binding of colesevelam to other drugs should be less problematic than that seen with the older BASs, it is probable that it too binds levothyroxine and decreases its absorption [137]. Therefore, caution should be taken when using colesevelam and levothyroxine together.
Other agents
Limited data are available on the potential impact of acarbose on thyroid function and thyroid cancer risk/benefit. Acarbose increased levels of TH in dexamethasone-induced hyperglycemic mice, although the mechanism of this is not well understood [138]. In a retrospective study, acarbose was associated with lower rate of papillary thyroid carcinoma growth [139].
THYROID DISORDERS AND HYPOGLYCEMIA
Glucose is an essential energy resource for the survival and activity of the brain. As the brain is unable to store glucose itself, it is very important that adequate plasma glucose levels be maintained and available to the brain at all times. There are several protective mechanisms to prevent or remediate hypoglycemia. A decrease in insulin secretion and activation of the counterregulatory response are the main response mechanisms for avoidance or correction of hypoglycemia. Counterregulatory hormones that act to prevent hypoglycemia include: glucagon, epinephrine, GH, cortisol, and neurotransmitters [142]. Hypoglycemia is common in T1DM and T2DM, in patients using insulin or some oral hypoglycemia agents. Hypothyroidism is one of the most common endocrine disorders, and frequently coexists with T1DM or T2DM [25,143]. Hypothyroidism may contribute to hypoglycemia via affecting various hormones and nervous function. In secondary hypothyroidism caused by hypopituitarism, hypoglycemia may be induced by adrenocorticotropic hormone and GH deficiency, but the risk of hypoglycemia is also increased in patients with primary hypothyroidism. TH affects hypothalamic-pituitary-adrenal (HPA) function and reduces GH and cortisol secretion, thereby interfering with the appropriate counterregulatory response to insulin induced hypoglycemia. Primary hypothyroidism reduces basal and stimulated GH, by acting on both the hypothalamus and pituitary [144]. In addition, hypothyroid patients may have co-existing relative adrenal insufficiency, thus weakening HPA response to hypoglycemia [145]. Hypothyroidism also affects glucose homeostasis, and these changes may be related to hypoglycemia. In hypothyroid patients, gluconeogenesis is reduced in skeletal muscle and in adipose tissue [146] and glycogenolysis is impaired [147]. These changes induce a delayed recovery from hypoglycemia. There are also reports that hypothyroidism may contribute to hypoglycemia through mechanisms including potentially impaired glucagon response and slowed gastric emptying [148,149].
CONCLUSIONS
TH affects glucose homeostasis, and thyroid disorders and DM are associated with each other. Autoimmunity is an important element in the relationship between T1DM and AITD. Thyroid dysfunction, both hyperthyroidism and hypothyroidism, is associated with insulin resistance and T2DM. Hypothyroidism can also increase risk of hypoglycemia in some setting. The association between TH and DM is also seen in pregnant women with gestational or pre-existing T1DM or T2DM. Screening for thyroid dysfunction is usually recommended in patients with T1DM, but there is a lack of recommendations in the screening for thyroid disease in patient with T2DM. Moreover, most guidelines do not recommend universal thyroid function screening in pregnant women, but instead limited to high-risk groups including T1DM. The use of certain antidiabetic agents influences thyroid function, TED, and thyroid cancer risk. Therefore, caution is required not only in interpreting TH levels when using these drugs in patients with T2DM, but also in using the drugs in patients with TED or risk of thyroid cancer. In conclusion, future research is warranted to further investigate the connections between DM and thyroid disorders and clinicians should take special consideration when evaluation patients with these conditions.
 XML Download
XML Download