

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The incidence of diabetes has been increasing rapidly for several decades worldwide. There were more than 400 million patients with diabetes in 2019, and this number is estimated to reach 700 million by 2045 [1]. In the Western Pacific region, including East Asia, there was an especially large increase in the number of diabetic patients after World War II because of dietary changes and a decrease in physical activity. East Asian individuals have lower insulin secretory capacity than Caucasians; thus, East Asians are considered to have a tendency to develop pancreatic β-cell failure owing to genetic factors [2-4].
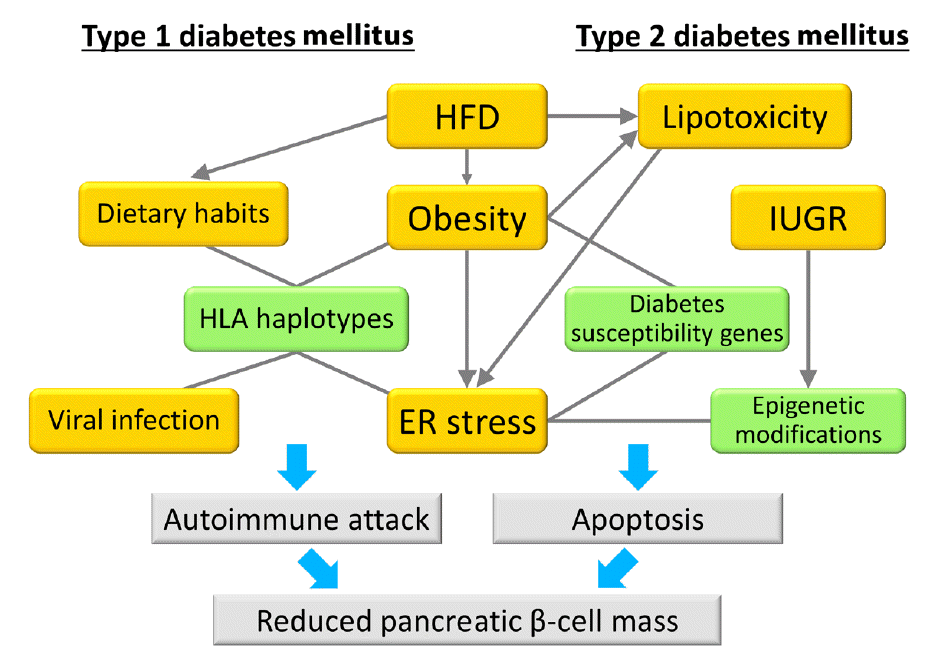
Previous work has shown the importance of insulin signaling in pancreatic β-cells using β-cell-specific transgenic mice. In 2006, Hashimoto et al. [5] clarified that a deficiency of 3-phosphoinositide dependent protein kinase 1 (Pdk1), an insulin signaling-related molecule, resulted in severe pancreatic β-cell failure, leading to hyperglycemia. In addition, it has been reported that mice deficient for tuberous sclerosis complex 2, a downstream molecule of Pdk1, exclusively in pancreatic β-cells develop hyperinsulinemia as a result of increased β-cell mass at younger ages, but show downregulation of insulin signaling induced by autophagic impairment and negative feedback, resulting in pancreatic β-cell failure at older ages [6-8]. To date, many investigators have used genetically modified mice as the main tool for the study of β-cell failure. These studies have revealed the pathogenesis of diabetes and the important role of various molecules in the onset of pancreatic β-cell failure. However, diabetes is a multifactorial disease. There are only a few monogenic forms of diabetes, such as maturity onset diabetes of the young, and it is considered that pancreatic β-cell failure as well as diabetes generally occur through a combination of genetic and environmental factors. The rapid increase in the number of diabetic patients in East Asia must be the result of genetic factors inherent in Asian people and environmental factors that have changed recently. Furthermore, for the combination of genetic and environmental factors, synergistic effects are considered to be more important than additive effects for the onset of diabetes and pancreatic β-cell failure. Thus, environmental factors increase the influence of some genetic factors synergistically, resulting in the development of diabetes and pancreatic β-cell failure (Fig. 1). Understanding gene-environment interactions is expected to lead to the personalization of diet and exercise therapy. In this review, we describe gene-environment interactions according to the findings of previous studies.
PHENOTYPIC VARIATIONS AMONG MOUSE STRAINS AND HUMAN ETHNIC GROUPS
First, we discuss the effects of diet in different mouse strains as a simple example of gene-environment interactions. Currently, C57BL/6J is the most widely used mouse strain in animal experiments because this strain readily shows phenomena such as obesity with a high-fat diet (HFD) [9,10]. C57BL/6J mice are lean with a normal chow diet, but they develop hyperglycemia, hyperinsulinemia, and hyperleptinemia with a HFD [11-13]. On the other hand, strains such as 129/Sv or A/J are resistant to obesity and hyperglycemia under a HFD [14]. When C57BL/6J and 129/Sv mice are fed an HFD, 129/Sv mice show an increase in thermogenesis and maintain insulin sensitivity. When the genetic backgrounds of both strains were compared by a genome-wide scan, there was a significant difference in a locus associated with insulin resistance located on chromosome 14 [15]. Namely, the genetic differences between these strains are considered to produce their divergent phenotypic responses to a HFD.
Such phenomena are also observed in humans, with differences in body mass index, energy consumption, insulin resistance, and insulin secretion between East Asian and Caucasian populations [16-20]. Moreover, it has been recently reported that the regulation of pancreatic β-cell mass varies among racial groups [21-24]. Saisho et al. [21] found that pancreatic β-cell mass was increased in an obese group of healthy Caucasians when compared with a lean group, which was considered to be due to a compensatory mechanism for insulin resistance. Conversely, when pancreatic β-cell mass was quantified in healthy Japanese subjects and compared between obese and lean groups, there was no significant difference in pancreatic β-cell mass between the groups, but there was a slight decrease in the obese group compared with the lean group [22]. These results indicate that the compensatory mechanisms for pancreatic β-cell mass under conditions of insulin resistance, such as obesity, are different between Caucasian and Japanese individuals. It is considered that the phenotype may differ according to genetic factors, even if the dietary habits are the same.
DEVELOPMENTAL ORIGINS OF HEALTH AND DISEASE
Environmental factors can affect genetic factors in adulthood, but the most important gene-environment interactions occur during the fetal period. In 1986, Barker and Osmond [25] proposed a theory that malnutrition in early life, including fetal life, causes susceptibility to ischemic heart disease and diabetes in adulthood (Barker’s hypothesis). This hypothesis, which is also referred to as the Developmental Origins of Health and Disease theory, suggests that the embryonic or neonatal environment significantly contributes to the onset of future metabolic diseases [26-30]. A 50-year epidemiological survey reported that children of mothers who experienced the “Dutch famine” at the end of World War II have a high incidence of hypertension, obesity, or glucose intolerance [31,32]. Furthermore, the mechanism underlying this phenomenon has been elucidated using animal models. Yura et al. [33] generated intrauterine growth restriction (IUGR) mice with uterine artery ligation as a model of undernutrition. They showed that IUGR mice have significantly lower birth weight than control mice, but there is no significant difference in body weight in adulthood between both groups because IUGR mice show “catchup” growth after weening. In addition, when both groups were fed a HFD, IUGR mice exhibit significantly more severe obesity and glucose intolerance [33]. This is considered to be due to a “neonatal leptin surge,” that is, an increase in serum leptin levels during the neonatal period, which is considered to lead to the onset of obesity by inducing leptin resistance.
Other reports have indicated that catch-up growth is associated with the development of obesity in later life in humans and mice [34-38]. Catch-up growth is considered to contribute significantly to the development of metabolic syndrome. It has previously been reported that catch-up growth occurs in pancreatic β-cell mass as well as the whole body in the adult period and insulin signaling has a key mechanistic role in this process [39,40]. In addition, the offspring of female rats lacking vitamin B12 have been reported to be susceptible to insulin resistance in adulthood, and the offspring of pregnant rats fed a low-protein diet also develop insulin resistance through the reduction of protein kinase C zeta (PKCζ) expression in skeletal muscle [41-43].
Recently, an increase in fat intake has become a severe problem in developed countries. The offspring of HFD-fed female monkeys exhibit increased expression of gluconeogenic enzymes and transcription factors in hepatic tissues, resulting in the development of nonalcoholic fatty liver disease [44]. Interestingly, the female offspring of male HFD-fed rats exhibit pancreatic β-cell failure and hyperglycemia [45]. Compared with control mice, the female offspring of HFD-fed fathers show a change in the expression of 642 genes in microarray analysis using pancreatic islets. These changes were considered to induce pancreatic β-cell failure.
As described above, many reports have described the effects of fetal and neonatal environments on genetic factors, leading to type 2 diabetes mellitus (T2DM) and obesity. The most wellknown molecular mechanism underlying this process is epigenetics, and we discuss the association between T2DM and epigenetics in the next section.
EPIGENETICS
Epigenetics is regarded as an important molecular mechanism in not only metabolic diseases but also various other diseases. Epigenetics is a system that changes gene expression via DNA or histone modifications without mutation of the nucleotide sequence [46-48]. As it is considered that environmental changes can affect gene expression, we should not forget this mechanism when we consider gene-environment interactions. In particular, changes in the intrauterine environment can have an impact on epigenetic modifications, and fetal nutritional conditions memorized in embryonic tissue have been found to lead to the development of various diseases in the future (metabolic memory) [49-51]. Park et al. [52] generated an IUGR rat model by ligation of the uterine artery, and these rats developed pancreatic β-cell failure. When they analyzed epigenetic modifications in isolated islets of IUGR rats, they found that the expression of pancreatic and duodenal homeobox 1 (Pdx1), a transcription factor, was decreased through an increase in DNA methylation and a change of histone modification at the Pdx1 promoter. The offspring of low-protein diet-fed female mice developed T2DM in adulthood, and there was a reduction of hepatocyte nuclear factor 4 alpha (HNF4α) expression in the pancreatic islets of those mice. This phenomenon was shown to be due to the inhibition of HNF4α promoter-enhancer interactions [53]. In addition, in stem cells isolated from cord blood of neonates with IUGR, changes of DNA methylation in multiple genes including HNF4α were found [54]. IUGR was predicted to influence the expression of various transcription factors, regardless of species or tissue.
The effects of IUGR have also been investigated in other insulin target organs [55,56], for instance, histone modifications at the glucose transporter type 4 (Glut4) promoter contribute to a reduction in Glut4 expression through the suppression of transcription in the skeletal muscle of IUGR rats [57]. Furthermore, in hepatocytes, Ehara et al. [58] clarified that the expression of glycerol-3-phosphate acyltransferase 1 was regulated by DNA methylation of its promoter. Interestingly, the expression of lipogenic genes is affected in the male offspring (F1) of IUGR mice, and even in hepatocytes of F2 mice. The expression of liver X receptor-alpha (Lxra) and DNA methylation at the Lxra promoter in hepatocytes of F2 mice has been reported to be altered significantly [59]. The fact that the effect of IUGR was passed on not only to children but also to grandchildren was a huge surprise. Possibly, the effects of the Dutch famine after World War II may still remain today.
TYPE 1 DIABETES MELLITUS AND GENE-ENVIRONMENT INTERACTIONS
Type 1 diabetes mellitus (T1DM) is induced through the destruction of pancreatic β-cells by the autoimmune response [60,61], and it is likely to occur during childhood [62]. However, a number of individuals develop T1DM in adulthood [63]. Two large epidemiological studies, Diabetes Mondiale (DIAMOND) and European Diabetes (EURODIAB), reported that the incidence of T1DM was increasing worldwide [64,65]. The reasons given for this included changes in environmental factors. Therefore, in this section, we describe the relationship between gene-environment interactions and T1DM.
Specific human leukocyte antigen (HLA) alleles are genetic risk factors for the onset of T1DM [66] and influence its incidence and age of onset [67,68]. The highest risk HLA haplotypes are DRB1*03:01-DQA1*05:01-DQB1*02:01 (known as DR3) and DRB1*04-DQA1*03:01-DQB1*03:02 (known as DR4). Individuals heterozygous for DR3/4 have an odds ratio (OR) of 16.59 for T1DM, while DR3/3 and DR4/4 homozygotes have ORs of 6.32 and 5.68, respectively [66]. These ORs are considerably higher than those of T2DM susceptibility genes. However, it is difficult to explain how the number of T1DM patients is increased by genetic factors alone. In recent years, the number of obese people has increased gradually worldwide [69-71]. In addition, the prevalence of overweight and obesity has risen in children with T1DM [72,73]. The increase in the number of obese subjects may be associated with the increase in the number of patients with T1DM. Interestingly, in Finland and Norway, which are two high-incidence T1DM countries, a rate of increase of T1DM has plateaued in recent years [74], indicating that this phenomenon is due to a plateau in the growth of obesity in both countries [75]. The ratio of obese subjects in a T1DM group was shown to be nearly equivalent to that in the general population or underwent a rapid increase [76]. In recent reports, the risk of developing T1DM was 63% higher in obese children than in lean children [77], and obesity increased susceptibility to insulin resistance, resulting in stress on β-cells and rendering them more vulnerable to an autoimmune attack [78].
Viral infection has been focused on as an important environmental factor of T1DM. It has been previously reported that enteroviruses, coxsackievirus B, cytomegalovirus, rotaviruses, and encephalomyocarditis virus contribute to the pathogenesis of T1DM [79,80]. In addition, dietary habits were found to be associated with the onset of T1DM as well as T2DM. Protein from cow milk and gluten are associated with an increase or decrease in the risk of developing T1DM [81-84].
The mechanisms associated with the destruction of pancreatic β-cells by environmental factors have not been elucidated fully [85]. One possibility is that endoplasmic reticulum (ER) stress contributes to the destruction of β-cells [86]. Inflammatory cytokines released by insulitis disrupt ER homeostasis, resulting in the induction of the unfolded protein response. During this adaptive phase, activation of the inflammatory response is predicted to play an important role in the pathogenesis of T1DM [87]. Furthermore, the fact that ER stress induces translational errors suggests that aberrant insulin produced by ER stress may become a target of the autoimmune response as an antigenic peptide [88].
INTERACTIONS BETWEEN DIABETES SUSCEPTIBILITY GENES AND HFD FEEDING
Finally, we discuss recent findings about gene-environment interactions in T2DM. In 2008, the potassium voltage-gated channel subfamily Q member 1 (KCNQ1) gene was identified as a T2DM susceptibility gene through large-scale single nucleotide polymorphism analysis of Japanese T2DM patients [89,90]. Subsequent reports from many facilities confirmed that there is an association between KCNQ1 mutations and reduced insulin secretion [91-94], but the mechanism for the onset of diabetes remains unclear. It was previously reported that decreased expression of the non-coding RNA Kcnq1ot1, which is expressed from within the Kcnq1 genetic region, increases the expression of cyclin-dependent kinase inhibitor 1C (Cdkn1c) through epigenetic modifications, resulting in reduced pancreatic β-cell mass [95]. Kcnq1 is an imprinted gene, and Kcnq1ot1 expression is decreased in pancreatic β-cells of mice only when a Kcnq1 genetic mutation is paternally inherited. As a result, pancreatic β-cell mass is decreased by enhancing Cdkn1c expression. These results may partially indicate that Kcnq1 mutation is an underlying pathogenic mechanism of T2DM. However, the OR of developing T2DM was only 1.4, suggesting that T2DM is not induced by genetic mutation alone. Therefore, focus shifted to the fact that there is a CCAAT motif in the Cdkn1c promoter region. As the CCAAT motif is a binding site for members of the CCAAT enhancer-binding protein (C/EBP) transcription factor family, it was hypothesized that binding of C/EBP family members to this motif may further enhance Cdkn1c expression. C/EBPβ expression was found to be enhanced in the islets of db/db mice, a T2DM mouse model, and reported that the accumulation of C/EBPβ increased vulnerability to ER stress and resulted in reduced β-cell mass [96,97]. In addition, recent studies revealed that C/EBPβ also accumulates in the pancreatic islets of HFD-fed mice, which may cause pancreatic β-cell failure through enhanced Cdkn1c expression and the induction of ER stress, and is considered to contribute to the development of T2DM.
Therefore, the influence of C/EBPβ accumulation in pancreatic β-cells was investigated when Kcnq1ot1 expression is decreased. A significant increase in fed blood glucose levels and a reduction in serum insulin levels were observed in mice with decreased Kcnq1ot1 expression and C/EBPβ overexpression exclusively in pancreatic β-cells, compared to control groups, which was accompanied by a significant decrease in pancreatic β-cell mass. This is presumed to be due to increased binding of C/EBPβ to the Cdkn1c promoter as a result of epigenetic modifications such as loosening chromatin structure under reduced Kcnq1ot1 expression. These results revealed that the synergistic effect of genetic factors (Kcnq1 mutation) and environmental factors (C/EBPβ accumulation) could induce markedly reduced pancreatic β-cell mass.
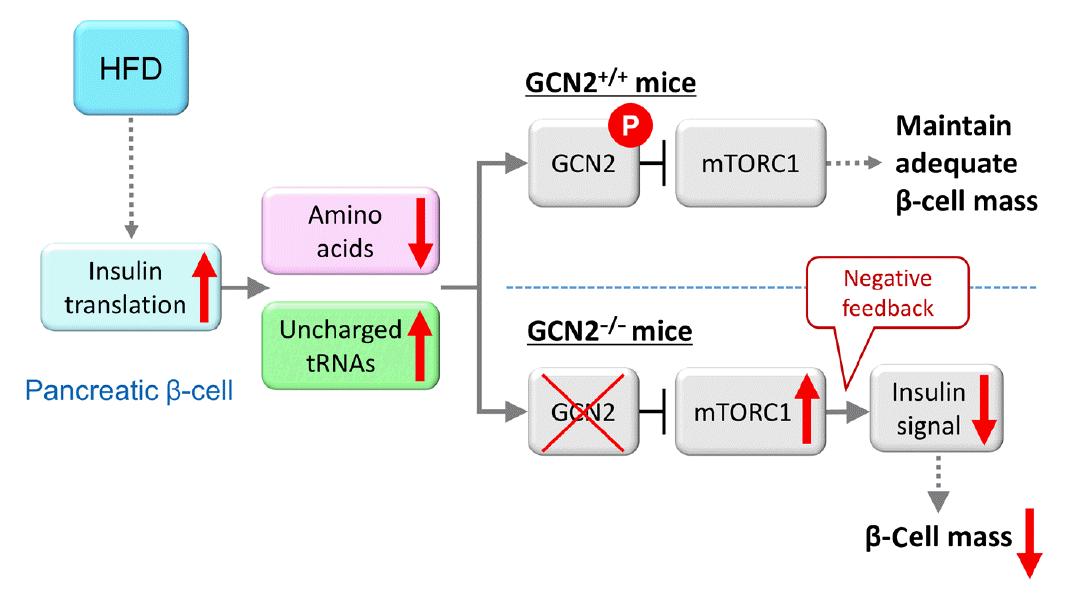
Furthermore, the role of eukaryotic translation initiation factor 2α kinase 4 (EIF2AK4) was investigated. EIF2AK4 encodes general control nonderepressible 2 (GCN2), which, in yeast and mammals, is activated as a result of amino acid deficiency and was identified as a T2DM susceptibility gene, in addition to KCNQ1, through large-scale single nucleotide polymorphism analysis of Japanese T2DM patients [98-101]. It was found that insulin secretion was reduced in carriers of the EIF2AK4 risk allele compared with individuals without the risk allele [102]. In addition, HFD-fed GCN2 knockout mice exhibit reduced pancreatic β-cell mass and decreased insulin secretion, while the phenotype of GCN2 knockout mice fed a normal chow diet is comparable to that of control mice [102]. This phenomenon was attributed to the constitutive enhancement of mechanistic target of rapamycin complex 1 (mTORC1) activity in pancreatic β-cells of GCN2 knockout mice. In fact, chronic enhancement of mTORC1 signaling was shown to result in reduced pancreatic β-cell mass [6-8], and this mechanism was considered to contribute to the development of reduced β-cell mass in HFD-fed GCN2 knockout mice. GCN2 is known to be activated under cellular amino acid deficiency [99], and it was found that amino acid concentration was decreased in the pancreatic β-cells of HFD-fed mice [102]. The HFD was shown to increase insulin demand in the whole body, and a large quantity of amino acids was consumed due to insulin production in pancreatic β-cells [103], resulting in reduced amino acid concentrations in these cells [102]. In pancreatic β-cells of HFD-fed GCN2 knockout mice, GCN2 cannot be activated in spite of amino acid deficiency. HFD-fed GCN2 knockout mice were considered to show hyperglycemia accompanied by reduced pancreatic β-cell mass through the constitutive enhancement of mTORC1 due to GCN2 inactivation (Fig. 2). The effect of GCN2 alone is not sufficient to induce pancreatic β-cell failure; however, environmental factors such as a HFD interact with GCN2 synergistically to cause pancreatic β-cell failure. As far as previous studies have shown, pancreatic islets were the only tissue in which cellular amino acid levels were decreased [102]. This result indicates that the response of pancreatic β-cells to HFD feeding is unique, and that gene-environment interactions may have an especially strong influence on the viability of pancreatic β-cells, as many T2DM susceptibility genes are associated with insulin secretion. A recent report showed that pancreatic β-cell death caused by gene-environment interactions could be examined using human pluripotent stem cells [104]. This method may help to clarify the associations between diabetes susceptibility genes and gene-environment interactions.
CONCLUSIONS
The explosive increase in the number of diabetic patients worldwide in recent years is mainly due to changes in the environment, given that there have been no significant or consistent changes in genetic factors. However, changes in environmental factors are likely to affect genetic factors. In this review, we documented the evidence for the relationship between gene-environment interactions and IUGR or diabetes susceptibility genes. However, gene-environment interactions are believed to be universally applicable to the contemporary pathology of diabetes, especially reduced β-cell mass. Therefore, further detailed analysis of these gene-environment interactions may suggest the key for personalized medicine. We look forward to further progress in this field in the future.
 XML Download
XML Download