

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diabetic peripheral neuropathy is one of the most common complications of both type 1 and type 2 diabetes [1], affecting 6%–51% of diabetic patients [2]. The spectrum of diabetic neuropathy is wide with many varieties. More than one cell type is affected at the same time, which results in disruptions of cellular interactions [3]. Many types of neurons, including pain fibers, motor neurons, and autonomic neurons are reported to be affected by this complication [4]. In addition to neurons, Schwann cells (SCs) lose their ability not only to supply energy during diabetes for both myelinated and unmyelinated axons but also to transfer toxic lipid species to the axons they have contact with [3]. Peripheral axons and SCs are dependent on each other for vitality and maintenance of the differentiated cellular phenotype. Thus, attempts to repair peripheral nerve damage in diabetic neuropathy should include approaches targeting both SCs and neurons [5].
Many models of diabetic neuropathy have been investigated for a better understanding of its mechanism and progress. In this regard, in vitro models can complement animal-based studies to study the intricate interaction of various types of cells in the pathogenesis of diabetic neuropathy as well as to find better treatments for this complication. In addition, in vitro models satisfy ethical issues, which is meaningful in respecting the 3Rs (Replacement, Reduction, Refinement) principle in animal use in research [6]. Furthermore, before translating the results into in vivo models, researchers have been required to demonstrate the effects of their concepts as much as possible in in vitro models [6]. Given that hyperglycemia is the main cause of diabetic neuropathy [7,8], some groups established in vitro models of diabetic neuropathy based on primary cells harvested from adult normal and diabetic rodents [9–11] or from rat embryos [12,13] and cultured them in appropriate growth media under hyperglycemic conditions. In order to overcome the limitations in the proliferation rate of primary cells, other groups used transformed cells such as SC [14,15], neuroblastoma [16,17], and pheochromocytoma (PC12) [18,19] cell lines. These monoculture-based models provide valuable data on cellular damage and morphological and behavioral changes of a specific cell type induced by high glucose. However, little is known about the concurrent effects of hyperglycemia on both SCs and neurons and about the neurite myelination under abnormal metabolic conditions. To address these unclear questions, we first examined how SCs and neurons are affected under hyperglycemia in single cultures. The co-cultures of SCs with sensory and motor neurons were then established to study the effect of hyperglycemia on the interaction of SCs with neurons. Insulin is widely known as a glucose homeostasis regulating hormone [20] and it has also been identified recently as a neurotrophic factor that plays a role in synthesizing the critical structural proteins of neurons [21]. Thus, we next investigated the effect of insulin on diabetic neuropathy neurons based on our model established in this study. The results showed that insulin promoted neurite extension and myelination, suggesting that insulin has a dual function on both SCs and neurons.
Go to : 
MATERIALS AND METHODS
1. Primary neurons culture
Primary sensory and motor neurons were isolated from E15 rat embryos obtained from pregnant Sprague Dawley (SD) rats (InVivos Pte Ltd, Singapore) in compliance with the ethical guidelines and approved by the Institutional Animal Care and Use Committee (Protocol No.: R14-1635) at the National University of Singapore. All efforts were made to minimize animal suffering and the number of animals in this study. The isolation procedure was carried out under sterile conditions. In brief, the pregnant SD rats were completely euthanized by CO2 gas. The E15 embryos were then collected and placed in a cold L15 medium (11415064, Gibco, Grand Island, NY). Spinal cords were dissected in an L15 medium. Dorsal root ganglia (DRG) were carefully cut off the spinal cords. The membrane of the spinal cord was then removed and the whole spinal cords were transferred to a small Petri dish containing L15 medium supplemented with 1% penicillin–streptomycin (10378016, Gibco) and cut into small pieces. Both sensory and motor neurons were dissociated by 0.05% trypsin/EDTA (25300120, Gibco). Then, the dissociated neurons were counted and seeded into 96-well plates coated with Poly-D-Lysine (50 µg/mL) (A3890401, Gibco) and Laminin (10 µg/mL) (23017-015, Invitrogen, Waltham, MA). Particularly, the cells were seeded at the density of 10,000 cells/well and 5,000 cells/well for single cultures and co-cultures with mouse SCs, respectively, in 96-well plates.
2. Hyperglycemia culture
In this study, SC medium (1701, ScienCell, Carlsbad, CA) and NeurobasalTM Medium (21103049, Gibco), with the respective basal glucose concentrations 5.5 mM and 25 mM, were used. The complete neurobasal medium was prepared by supplementing 2% B-27TM Supplement (50X) (17504044, Gibco) and 0.5 mM GlutaMAXTM Supplement (35050061, Gibco). The complete SC medium was prepared by adding 25 mL of fetal bovine serum (FBS), 5 mL of Schwann Cell Growth Supplement, and 5 mL of penicillin/streptomycin solution. Hyperglycemic media was prepared by adding glucose (47829, Sigma-Aldrich, Singapore) to these complete media. In particular, the glucose level of the SC medium was adjusted to 10, 30, and 60 mM and that of the Neurobasal Medium was adjusted to 35, 45, and 60 mM to produce hyperglycemic insults. These media were used at the cell time of seeding. For culture experiments lasting for more than 2 days, media was changed every 2 days to maintain the hyperglycemic condition during the experiments. For co-culture studies, SCs (ScienCell) were seeded into a 96-well plate at the density of 5,000 cells/well (for coculture in hyperglycemic insults alone) or 3,000 cells/well (for coculture in hyperglycemic insult supplemented with insulin) and induced to the mature phase in the complete SC medium supplemented with vitamin C (50 μg/mL) for 4 days prior to seeding neurons on top of the SC layers. The complete Neurobasal Medium with the assigned glucose concentrations was used for co-culture after seeding the neurons. All hyperglycemia cultures were performed in a randomized manner.
3. Insulin treatment
For the single culture of neurons, insulin (I9278, Sigma-Aldrich, Saint Louis, MO) ranging from 0.01–2 µM was introduced to the complete neurobasal medium at the seeding time and incubated for 2 days. For the experiment evaluating the effect of insulin on myelination, the treatment was carried out in a blinded manner. The complete neurobasal medium (adjusted to 60 mM glucose) supplemented with or without insulin at a dose of 0.1 µM was used after seeding neurons on top of the SC layers and replaced every two days of culture.
4. Immunocytochemistry
After removing the culture media, the cells were rinsed by PBS 3 times for 3 minutes each time. The cells were then fixed using 4% paraformaldehyde (158127, Sigma-Aldrich, Singapore) for 30 minutes at room temperature (RT), followed by PBS rinsing. After that, the cells were incubated with blocking solution containing 3% BSA (A7906, Sigma-Aldrich, Singapore) and 0.25% TritonX-100 (X100, Sigma-Aldrich, Singapore) for 1 hour at RT. Then, the cells were incubated overnight at 4°C with the appropriate primary antibodies. In particular, mouse anti-MBP (ab62631, 1:500) and rabbit anti-p75NGFR (ab227509, 1:500) antibodies were applied for the single culture of SCs. For the single culture of neurons, mouse anti-NF160 antibody (ab7794, 1:500) was used. For the co-culture of SCs and neurons, mouse anti-NF160 and rabbit anti-MBP (ab40390, 1:200) antibodies were used. Anti-MBP antibodies react with myelin basic proteins that are common participants in myelin sheaths. Anti-p75NGFR reacts with nerve growth factor receptor found on both neuronal cells and glial cells. In glial cells, p75NGFR is a positive modulator for myelination during development. Anti-NF160 antibody reacts with both phosphorylated and non-phosphorylated forms of medium neurofilament protein. All primary antibodies were purchased from Abcam (Cambridge, UK). After washing by PBS, the cells were incubated for 1 hour at RT with Alexa 488 goat anti-mouse (A-11017, 1:1,000, Molecular Probes, Eugene, OR) and Alexa 546 goat anti-rabbit (A-11072, 1:500, Molecular Probes) as the secondary antibodies. After that, the cells were washed by PBS 3 times, each time for 3 minutes.
5. Live and dead assay
The effect of hyperglycemia on the survival of SCs was assayed at 7 days of culture in control medium (5.5 mM glucose) and hyperglycemic media (10, 30, and 60 mM) by LIVE/DEAD® Viability/Cytotoxicity Kit for mammalian cells (L3224, Molecular Probes). The protocol was performed according to the instruction of the manufacturer. In brief, the media culture was discarded and the cells were washed gently by PBS to remove FBS remaining in the cell culture dish. The working solution was prepared by diluting calcein AM and EthD-1 in SC medium to the final concentrations of 2 and 4 µM, respectively. A sufficient working solution was added into the experimental wells so that all cells were covered with solution. The well plate was then covered by aluminum foil and transferred to the incubator for 30 minutes. After removing the working solution, the cells were washed twice by PBS and the images were obtained with a laser scanning confocal microscope (LSM 800; Carl Zeiss, Jena, Germany). Each well was divided into five areas: top, bottom, left, right, and center areas. Two photos were acquired randomly at each area.
6. SCs proliferation assay
The proliferation rate of SCs under hyperglycemic culture was evaluated by alamarBlue® (AB) (BUF012B, Bio-Rad, Hercules, CA) staining at 1, 3, 5, and 7 days of culture. SCs were seeded into a 96-well plate at a density of 1,000 cells/well and were grown in SC medium containing 5.5 mM (control), 10 mM, 30 mM, and 60 mM of glucose at the seeding time. After removing the culture medium, the cells were washed three times by PBS (–). AB was then diluted by FBS-free SC medium (1:10) and added into the experimental wells (250 µL each well). After that, the culture plate was protected from light and transferred into the cell incubator for 2 hours. Then, the incubated AB of each well was transferred to 4 wells of a new 96-well plate (50 µL each well) for measurement of the fluorescence density using a microplate reader at the excitation and emission wavelengths of 570 nm and 600 nm, respectively. The number of viable cells correlates with the percentage of AB reduction that was calculated according to the manufacturer’s instruction:
In the formula, εOX and εRED are constants representing the molar extinction coefficient of AB oxidized form (BLUE) and of AB reduced form (RED), respectively. A and A’, respectively, represent the absorbance of test wells (at λ1 = 570 nm and λ2 = 600 nm) and negative control wells that contain SC medium and AB without cells.
7. Image acquisition and analysis
All immunofluorescent images were acquired on a laser scanning confocal imaging system LSM 800 (Carl Zeiss).
For evaluation of the de-differentiation of SCs under hyperglycemia, area, mean fluorescence, and several adjacent background readings of MBP- and p75NGFR-single channel of each image were first measured by FIJI (https://fiji.sc/). Then, the total corrected cellular fluorescence (TCCF) was calculated by a formula referred to in a previous paper by McCloy et al. [22]: (TCCF) = integrated density – (area of selected cell × mean fluorescence of background readings). Each well was divided into five areas: top, bottom, left, right, and center areas. Two photos were acquired randomly at each area.
For measurement of neurite length, Simple Neurite Tracer, which is an open plugin of FIJI, was used as described previously by More et al. [23]. At least 4 wells per group were analyzed. In the co-culture of SCs and neurons, the neurites that were positive for both NF-160 and MBP antibodies were considered myelinated neurites. The ones that were only positive for NF-160 antibody were counted as unmyelinated neurites. All neurons that were positive for NF-160 in each well were taken and analyzed.
8. Statistical analyses
All experiments were carried out in at least triplicate. The results were analyzed using one-way non-repeated ANOVA followed by Dunnett’s post hoc test (Figs. 1-5), one-way non-repeated ANOVA followed by Tukey’s post hoc test (Fig. 6), and a two-tailed Students t-test (Fig. 7). All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, San Diego, CA). The data were expressed as the mean ± standard error. P < 0.05 was considered significant.
 | Fig. 1Schwann cell proliferation is attenuated in culture under hyperglycemia. (A) Representative images show changes in cell morphology and confluence under hyperglycemic culture. Scale bar: 300 µm. (B) Representative images of live and dead assay performed at 7 days of culture in control and hyperglycemic conditions. Scale bar: 100 µm. (C) The graph shows the number of cells at 1, 3, 5, and 7 days of culture in control medium (5.5 mM glucose) and in hyperglycemic medium containing 10, 30, and 60 mM glucose. At 1 day, n = 4 wells each group; non-repeated-measures ANOVA, not significant. At 3 days, n = 4 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 12) = 6.327, **P < 0.01 vs. control group. At 5 days, n = 6 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 20) = 16.09, *P < 0.05, **P < 0.01, and ****P < 0.0001 vs. control group. At 7 days, n = 7 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 24) = 24.47, ****P < 0.0001 vs. control group. (D) The graph shows the percentage of dead cells at 7 days of culture in control medium (5.5 mM glucose) and in hyperglycemic media. n = 5 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 16) = 43.92, *P < 0.05 and ****P < 0.0001 vs. control group.
|
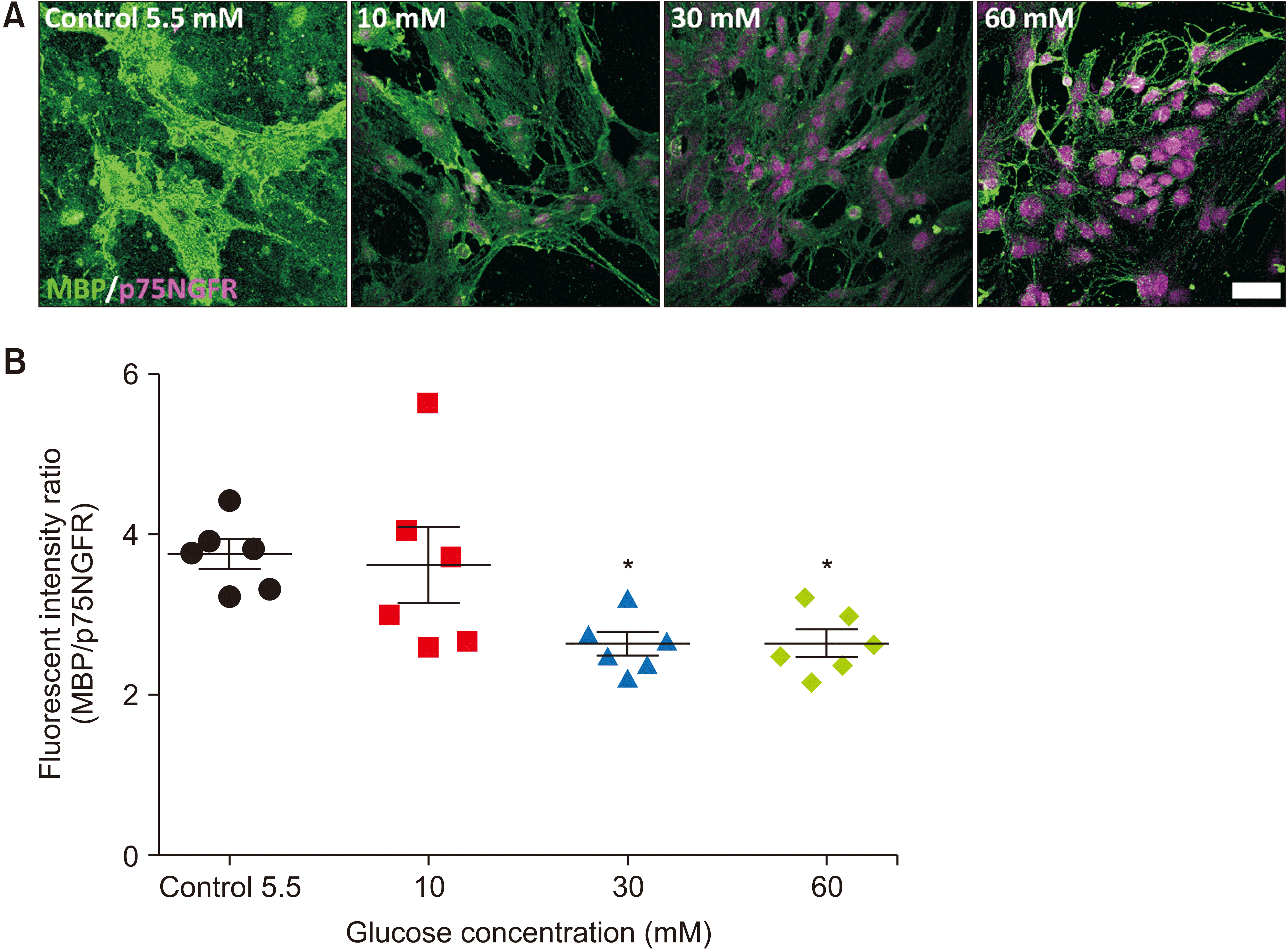 | Fig. 2Schwann cell maturation is impaired under hyperglycemia. (A) Representative images of immunocytochemistry with p75NGFR and MBP performed at 7 days of culture in control and hyperglycemic conditions. Scale bar: 50 µm. (B) The graph shows the fluorescent intensity ratio of MBP and p75NGFR at 7 days of culture in control medium (5.5 mM glucose) and in hyperglycemic medium containing 10, 30, and 60 mM glucose. n = 6, *P < 0.05 vs. control group. n = 6; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 20) = 4.845, *P < 0.05 vs. control group.
|
 | Fig. 3Hyperglycemia impairs neurite extension. (A, B) Representative images of sensory (A) and motor (B) neurons stained for NF-160 at 2 days of culture in control and hyperglycemic conditions. Scale bar: 50 µm. (C, D) The graph shows the significantly decreased average neurite length of sensory (C) and motor (D) neurons under hyperglycemia at 2 days of culture. (C) n = 3 wells each group, non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 8) = 32.57, ***P < 0.001 vs. control group. (D) n = 3 wells each group, non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 8) = 22.14, **P < 0.01 and ***P < 0.001 vs. control group.
|
 | Fig. 4Hyperglycemia impairs myelination of sensory neurites. (A) Representative images of sensory neurons stained for NF-160 and MBP at 7 days of co-culture with Schwann cells in control and hyperglycemic conditions. Scale bar: 100 µm. (B) The graph shows the significantly decreased percentage of myelinated sensory neurites under hyperglycemia. n = 3 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 8) = 50.10, **P < 0.01 and ****P < 0.0001 vs. control group.
|
 | Fig. 5Hyperglycemia impairs myelination of motor neurites. (A) Representative images of motor neurons stained for NF-160 and MBP at 7 days of co-culture with Schwann cells in control and hyperglycemic conditions. Scale bar: 100 µm. (B) The graph shows the significantly decreased percentage of myelinated motor neurites under hyperglycemia. n = 3 wells each group; non-repeated-measures ANOVA followed by Dunnett’s post hoc test, F (3, 8) = 238.5, ****P < 0.0001 vs. control group.
|
 | Fig. 6Insulin promotes neurite extension. (A, B) Representative images of sensory (A) and motor (B) neurons stained for NF-160 at 2 days of culture in neurobasal medium (25 mM glucose) supplemented with a series of insulin concentrations ranging from 0.01–2 µM. Scale bar: 50 µm. (C) The graph shows the significantly different average neurite length of sensory neurons under the effect of insulin. n = 3 wells each group; non-repeated-measures ANOVA followed by Tukey’s post hoc test, F (6, 14) = 3.970, *P < 0.05 vs. control group. (D) The graph shows the significantly different average neurite length of motor neurons under the effect of insulin. n = 3 wells each group; non-repeated-measures ANOVA followed by Tukey’s post hoc test, F (6, 14) = 47.26, ****P < 0.0001 vs. control group; #P < 0.05 and ##P < 0.01 vs. 0.01 µM; ιιιP < 0.001 and ιιιιP < 0.0001 vs. 0.05 µM; aP < 0.0001 vs. 0.1 µM.
|
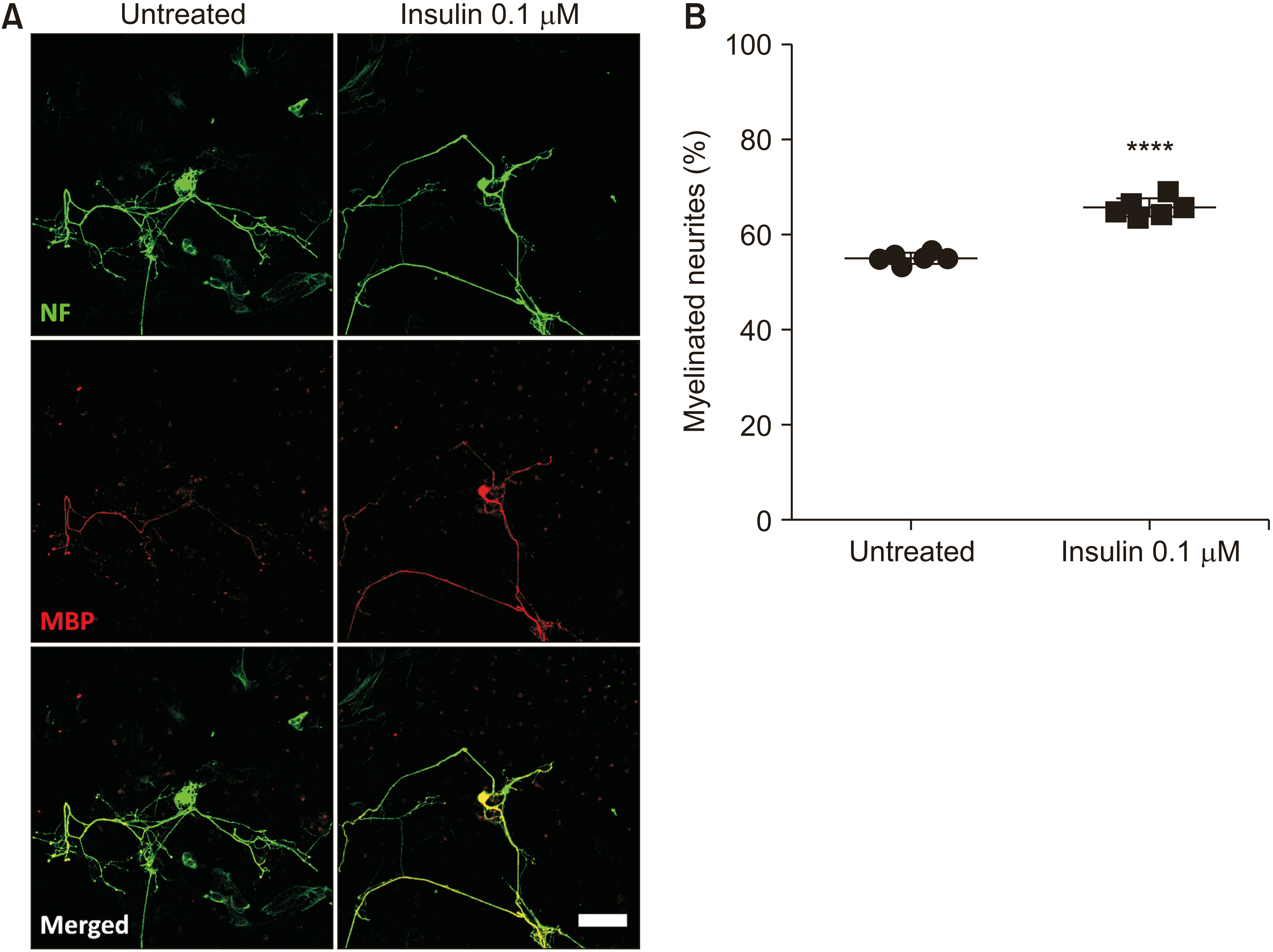 | Fig. 7Insulin promotes sensory neurite myelination. (A) Representative images of sensory neurons stained for NF-160 and MBP at 7 days of co-culture with Schwann cells in hyperglycemic insult (60 mM glucose) supplemented with insulin at 0.1 µM. Scale bar: 100 µm. (B) The graph shows the significantly promoted percentage of myelinated sensory neurites in the group treated by insulin. n = 6; two-tailed Student’s t-test, t (4) = 8.135, ****P < 0.0001.
|
Go to :
RESULTS
1. Hyperglycemia attenuates the proliferation of SCs
Unlike neurons, SCs can divide indefinitely throughout life [24]. They have no ability to transmit synaptic messages; however, they play important roles in myelinating axons, guiding neurons, and eliminating cellular debris [25,26]. In this study, given that hyperglycemia, a key contributor to the progress of diabetic neuropathy [8], attenuates the proliferation of SCs, we evaluated the proliferation rate of SCs under hyperglycemic culture. Hyperglycemia was simulated by adjusting the glucose concentration of the SC medium from the basal level 5.5 mM to 10, 30, and 60 mM. These modified media were introduced to SCs at the seeding time and changed every two days to maintain the hyperglycemic condition during the experiments. The cells grown in hyperglycemic insults also presented decreased confluence (Fig. 1A, B). The proliferation of SCs was then assayed by alamarBlue staining at 1, 3, 5, and 7 days of culture under the hyperglycemic condition and compared with that in the normal medium. No major change in the cell density was found in any group after 1 day of culture. However, the cell proliferation in high glucose groups was significantly decreased by 3, 5, and 7 days of culture compared to the control group. Particularly, by 7 days, the number of cells in the control group was 3,076 ± 32.47 vs. 2,470 ± 72.57 (F3, 24 = 24.47, P < 0.0001) and 2,162 ± 53.30 (F3, 24 = 24.47, P < 0.0001) in the 30 mM and 60 mM, respectively (Fig. 1C).
In addition, the viability of cells was examined by the LIVE/DEAD® Viability/Cytotoxicity Assay Kit at 7 days. As shown in Fig. 1B, live cells and dead cells fluoresced bright green and red-orange, respectively. In the basal level of glucose, the cells showed a higher density, and only a few dead cells (1.28 ± 0.19%) were found. In contrast, the cells grown in hyperglycemia media had a low density and a significantly higher percentage of dead cells (2.58 ± 0.54%, 3.57 ± 0.59%, and 8.61 ± 0.51%, respectively, in the 10, 30, and 60 mM glucose-treated groups) (F3, 16 = 43.92, P < 0.05 control vs. 30 mM group, P < 0.0001 10 mM vs. 60 mM group).
These results showed that hyperglycemia significantly attenuated the proliferation and viability of SCs.
2. Impaired differentiation capacity of SCs under hyperglycemic insults
SC myelination plays a crucial role not only in regulating peripheral nerve function and conduction velocity but also in maintaining the architecture of axons [9,27]. In diabetic neuropathy, loss of myelinated fibers is one of the most common reported histologic changes [28]. In addition, previous studies showed that hyperglycemia triggers abnormal signaling pathways in the proliferation and differentiation of SCs [9,10]. In this study, to examine the changes in the differentiation of SCs under hyperglycemia, we analyzed the expression of immature (p75NGFR) and mature markers (MBP) of SCs at 7 days of culture under hyperglycemia. The ratio of fluorescent intensity of MBP and p75NGFR in the control group (3.748 ± 0.1774) was significantly higher than that in the high glucose concentration groups (2.643 ± 0.1433, F3, 20 = 4.845, P < 0.05 and 2.638 ± 0.1618, F3, 20 = 4.845, P < 0.05 in the 30 and 60 mM glucose groups, respectively) (Fig. 2), suggesting that hyperglycemia impaired the maturation of SCs.
3. Hyperglycemia attenuated neurite extension
Unlike other cell types in our body, neurons have a unique morphology with axons that extend their branches over long distances to form connections with other cells [29]. In addition, neurons have dendrites that play an important role in receiving input from other neurons and cells in the environment [30]. Any abnormalities in the morphology of neurons will result in reduced neuronal activity and increased neurodegeneration. The biological properties, especially neurite outgrowth, were previously reported to be altered under diabetic conditions [12].
To examine the morphological changes of neurons cultured under conditions mimicking hyperglycemia, we measured the average neurite length of neurons by using an open plugin of FIJI named Simple Neurite Tracer, as described previously by More et al. [23]. As shown in Fig. 3, the average neurite length of both sensory and motor neurons cultured in media containing high concentrations of glucose was significantly shorter than that in the control group. The average neurite length of the sensory neurons in the control group was 2,179.9 ± 106.7 µm, which was significantly longer than in the 45 mM- (1,655.5 ± 43.2 µm, F3, 8 = 32.57, P < 0.001) and 60 mM-glucose treated (1,457.8 ± 126.0 µm, F3, 8 = 32.5, P < 0.001) groups (Fig. 3C). A significant difference was also found in control motor neurons (2,687.1 ± 141.0 µm) vs. the 45 mM group (2,190.4 ± 101.1 µm, F3, 8 = 22.14, P < 0.01) and 60 mM group (1,828.4 ± 104.6 µm, F3, 8 = 22.14, P < 0.001) (Fig. 3D). These in vitro results were consistent with those reported in diabetic animal models [31–33].
4. Impaired neurite myelination under hyperglycemia
Formed by SCs in the peripheral nervous system and oligodendrocytes in the central nervous system, myelin is a unique plasma membrane with high lipid content (~70%). Myelin plays a crucial role in accelerating conduction velocity for the rapid conductivity of action potentials along neurons [34,35]. Our results first show that the proliferation and differentiation of SCs are impaired under diabetic conditions (Figs. 1, 2), suggesting that the neurite myelination by SCs will also be affected during diabetes. Indeed, our results showed that compared with the control groups, both sensory and motor neurons co-cultured with SCs under hyperglycemia showed an attenuated percentage of myelinated neurites. In the sensory neuron-SC co-culture, compared to the control group (90.35 ± 0.72%), the percentage of myelinated neurites was significantly impaired in the 35 mM group (85.86 ± 0.39%, F3, 8 = 50.10, P < 0.01) and further reduced in the 45 mM (F3, 8 = 50.10, 83.39 ± 0.72%, P < 0.0001) and the 60 mM (F3, 8 = 50.10, 80.92 ± 1.63%, P < 0.0001) groups (Fig. 4). The percentage of myelinated neurites was much lower than that in all groups of the motor neuron-SC co-culture system; however, this result was in accordance with the previous observation of Hyung et al. [36]. Particularly, the myelination of motor neurons proceeds through two distinct stages. In the pre-myelinating stage, expression of MBP in SCs was mostly widespread for 10 days in vitro. In contrast, SCs in the myelinating stage are tightly localized along the axons and reached 80% by 21 days in vitro [36]. Nonetheless, the significantly decreased myelination of motor neurons was also found in all groups cultured under hyperglycemia conditions compared to the control group (Fig. 5). Thus, hyperglycemia significantly impaired the myelination of SCs to both sensory and motor neurons.
5. Insulin enhanced neurite extension
To examine the effect of insulin on neurite extension, a series of insulin solution ranging from 0.01–2 µM was supplemented to the culture media of primary neurons at the seeding time. At two days of culture, neurons were stained with NF-160, and neurite length was measured as described above. In the sensory neurons culture, insulin remarkably increased the neurite length at all examined concentrations, with significant enhancement found at the concentrations 0.01 (F6, 14 = 3.970, P < 0.05), 0.05 (F6, 14 = 3.970, P < 0.05), and 0.1 µM (F6, 14 = 3.970, P < 0.05). At 0.5–2 µM, the neurite length tended downward. In contrast, insulin promoted neurite length of motor neurons at only 0.01 µM (no significance found). The neurite length was impaired at 0.05 µM and was further impaired thereafter (Fig. 6B). Interestingly, we found that at 2 µM, motor neurons formed clusters, suggesting that insulin may support migration of motor neurons.
6. Insulin promoted neurite myelination of diabetic neuropathy neurons
Based on the results of the in vitro model of diabetic neuropathy established above, we used the media adjusted to 60 mM glucose to further examine the effect of insulin on promoting the myelination of SCs to diabetic neuropathy neurons. Our results in Fig. 4 showed that hyperglycemia attenuated sensory myelination. However, we noticed that at the density of 5,000 SCs/well, the percentage of myelinated neurites in the 60 mM group reached 80.92 ± 1.63% (Fig. 4B). To examine the potential effect of insulin on myelination, we therefore decreased the number of seeded SCs to 3,000 cells/well. In addition, the results in Fig. 6 revealed that insulin at 0.1 µM showed the highest level of neurite length promotion. As a result, we also examined the effect of insulin at this concentration on neurite myelination. The coculture of SCs and sensory neurons was described in the Materials and Methods section. Insulin was introduced to the media culture at the cell seeding time.
The results showed that compared to the untreated group (55.86 ± 1.37%), insulin significantly enhanced neurite myelination to 63.33 ± 0.78%, t (4) = 8.135, ****P < 0.0001. The results suggest that insulin may play a dual role in the improvement of diabetic neuropathy by promoting both neurite extension and myelination. In contrast to sensory neurons, however, the signal of myelin protein observed in motor neurons at 7 days co-cultured with SCs was low (data not shown), suggesting that more culture time is needed to examine the effects of insulin on motor myelination.
Go to :
DISCUSSION
Many animal models of diabetic neuropathy have been successfully established by us, with a neuropathy complication developed from type-2 non-obese diabetes [31], and by others [37–40]. These models showed remarkable contributions to studies on the mechanism and progression of the neuropathy complication of diabetes and elucidated approaches for the treatment of this complication. However, the onset and progression of neuropathy in these animal models has not been totally understood [41]. Understanding of the mechanisms that trigger diabetic neuropathy at the cellular and molecular levels still remains unclear [3]. Thus, researchers have used combined in vivo and in vitro models of diabetic neuropathy in their attempts to better understand the complex mechanisms of this complication.
In this regard, findings achieved from in vitro model studies can complement animal research and suggest different aspects to investigate. In addition, in vitro models have advantages over animal models, including lower cost, ease of use and control, and a higher experimental throughput. More importantly, in vitro models can offer a simplified platform for examining new treatment approaches, such as pharmacological screening [42]. in vitro models of diabetic neuropathy based on co-cultures are advantageous in addressing questions regarding the spectrum of diabetic neuropathy and the interplay of surrounding cells. Particularly, it was noticed that approaches that will support both SCs and neurons should be considered equally for attempts to repair peripheral nerve injuries [5]. In this study, given that hyperglycemia is the main cause of diabetic neuropathy [8], we first investigated the most appropriate glucose concentration to establish the model in single cultures of SCs, sensory, and motor neurons. We then established the co-culture system of SCs with sensory and motor neurons to study the effect of hyperglycemia on myelination. Our results were consistent with those that have been reported in animal models of diabetic neuropathy, especially increased apoptotic neurons and SCs (data not shown) [43], impaired neurite extension [31], and impaired myelination [3].
The difference in the vulnerability level between sensory and motor neurons to diabetes-mediated injury has been noticed in both human diabetic patients and animal models [3,44]. Particularly, sensory neurons are more vulnerable [3,44] and are involved in earlier stages of diabetes [45]. This notion was also observed in our non-obese diabetes model. Seventeen weeks after co-injected by streptozotocin and nicotinamide, mice showed impaired sensory nerve conduction and the number of intraepidermal nerve fibers was significantly decreased. In contrast, impairment of the innervation of motor neurons was not found [31].
Several reasons have been proposed to explain why sensory neurons are more vulnerable to diabetic neuropathy. Hameed [46] reported that sensory axons express more distinct voltage gated sodium channels (Nav1.6, 1.7, 1.8, and 1.9) with different biophysical characteristics compared to those in motor axons (Nav1.6). The expression of these sodium channels possesses distinct electrophysiological properties in sensory axons. Specifically, Nav1.7 and 1.9 produce a persistent sodium current near the resting membrane potential. In addition, due to their smaller diameter than motor axons, sensory axons have a higher surface area to volume ratio, resulting in changes in sodium and calcium current and rendering sensory axons more vulnerable to injury [3]. Furthermore, sensory axons are more susceptible to diabetes because of their physiological environment. Sensory neurons are protected by the blood–ganglion barrier, which is more permeable to macromolecules and other constituents than the blood–nerve barrier that protects motor neurons [44]. Thus, DRG sensory neurons are more easily exposed to blood-borne toxins and more dependent on trophic support provided by SCs which are also remarkably affected by diabetes. Sensory neurons also have higher metabolic demand, supported by local blood flow, leaving them at greater risk from diabetic nutrient blood flow [44]. In the present in vitro study, we also found the further impairment of sensory neurite length compared to that of motor neurons. In particular, at 45 mM and 60 mM, the neurite length of sensory neurons reduced by 24.04% and 33.01%, respectively. However, compared with the corresponding hyperglycemic insults, the reduced percentage of neurite length in motor neurons was 18.49% and 31.96% (Fig. 3). In addition, the percentage of myelinated neurites between sensory and motor neurons was also noticed and discussed above. We also found different sensitivity levels to insulin between sensory and motor neurons; however, further studies are needed to elucidate the mechanism.
Insulin, a member of the insulin-like superfamily, in addition to being widely known as a glucose homeostasis regulating hormone, has been identified recently as a neurotrophic factor that plays a role in synthesizing critical structural proteins of neurons [21]. In diabetes, the insulin signaling is disrupted due to insulinopenia (type 1) or insulin resistance (type 2) [47], as well as insulin deficiency found in both type 1 and type 2 diabetes [48] that may lead to a delay in nerve regeneration [31]. Previous studies demonstrated that insulin receptors (IRs) are expressed in many neural cell types such as SCs [49] and DRG [7]. Some interesting questions and hypotheses have been raised about the role of IRs in neural cells because the majority of neurons are not dependent on insulin to take up glucose [21]. Furthermore, the expression of IR in SCs during development parallels the myelin structural protein P0 expression (at both mRNA and protein levels) and the growth of the myelin sheath [50]. These observations suggest that insulin may play a role in myelination and peripheral nerve support. In this study, the authors focused on examining the effect of insulin on myelination in their established in vitro model of diabetic neuropathy based on a co-culture of SCs and neurons. The results showed that insulin remarkably promoted the percentage of myelinated fibers. In addition, insulin enhanced the neurite extension, suggesting that insulin can play a dual role in both promoting neurite extension and enhancing neurite myelination. However, at high concentrations (0.5 µM and 0.05 µM thereafter to sensory and motor neurons, respectively), insulin inhibited neurite extension, even though the number of surviving neurons was significantly higher than that in the untreated group or the groups treated with lower insulin concentrations (data not shown). More studies are required to draw conclusions regarding this observation. Combined with the authors’ other results regarding the effects of insulin on the proliferation and maturation of SCs (data not shown), insulin may be a potential candidate for improving diabetic neuropathy. There are suggestions for future works based on our findings. First, as a molecule mediating its role via two main pathways including AKT/mTOR and ERK/MAPK pathways, there are interactions between insulin and other molecules in their actions that may ultimately result in the promotion of myelination. Further studies are needed to discover the full potential of insulin alone or its combinative effects with other molecules in improving diabetic neuropathy.
In conclusion, the in vitro model of diabetic neuropathy based on the co-culture of SCs and neurons shows itself to be useful for various purposes, including studying mechanisms and examining new approaches to many cellular targets for the treatment of this complication. In regard to treatment, insulin showed the effects on the promotion of neurite extension and myelination, suggesting further studies in the future to discover the full potential of insulin in improving diabetic neuropathy.
Go to :
 XML Download
XML Download