

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) is an autosomal recessive inherited disease that disrupts genes encoding protein transporters responsible for bile formation [1]. Clinical manifestations in PFIC patients include progressive jaundice, pruritus, and growth retardation. Subsequently, patients will develop liver cirrhosis and hepatic failure. Liver transplantation (LT) has been regarded as the only effective treatment for PFIC.
In infant patients, hepatocellular carcinoma (HCC) is very rare. Most HCCs arise in the setting of prior liver disease. HCCs may be discovered incidentally in the explant or detected during surveillance of underlying disease, and it will trigger the decision to perform LT [2].
We herein present a case of living donor liver transplantation (LDLT) in a 7-month-old infant with PFIC and HCC using a left lateral section graft. The patient has been doing well for 8 years uneventfully.
CASE REPORT
This study was approved by the Institutional Review Board of Asan Medical Center (IRB No. 2020-0836), which waived the requirement for informed consent due to the retrospective nature of this study.
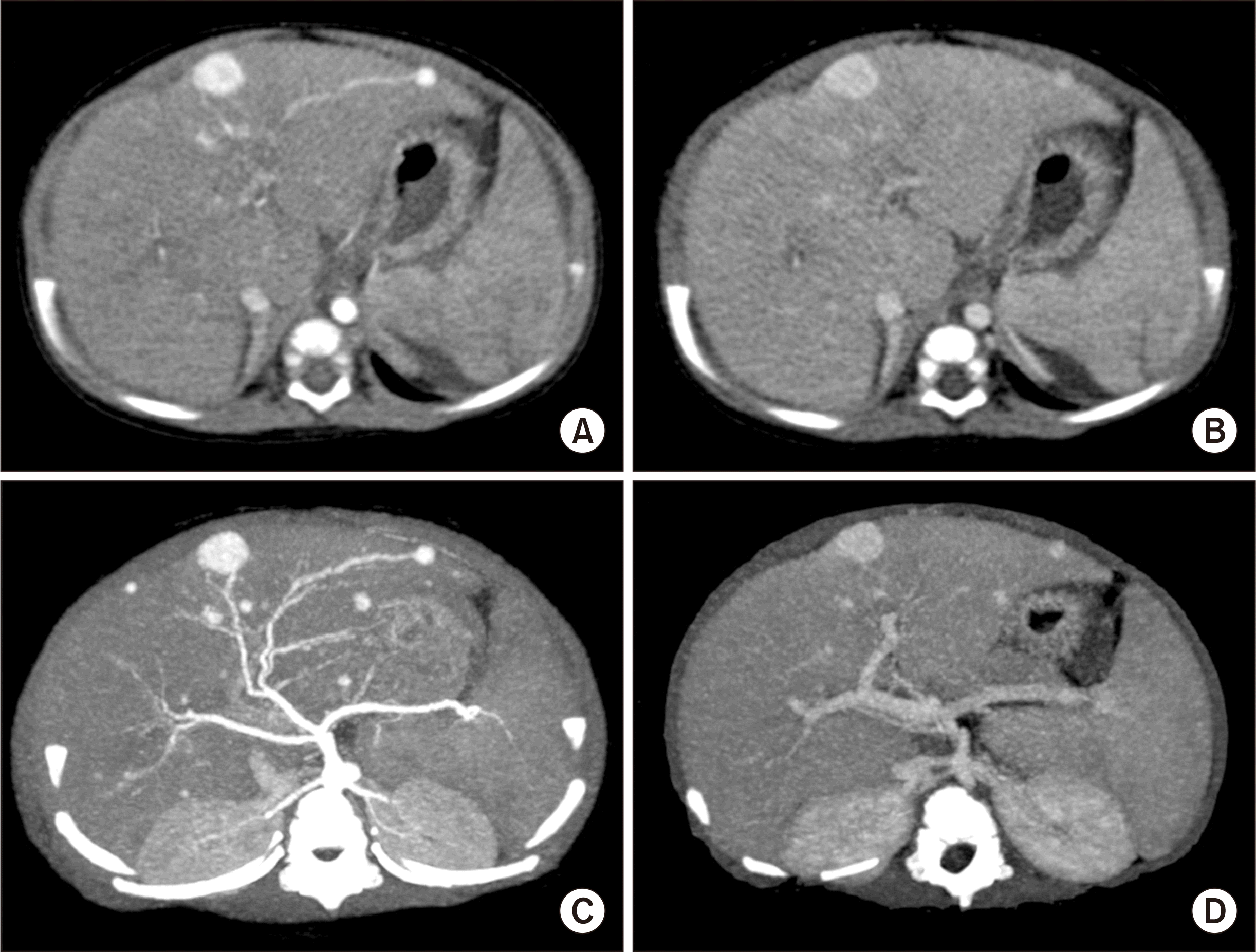
A 7-month-old female infant was referred to our institution for progressive jaundice. The patient was born through a full-term cesarean section delivery. Jaundice occurred after birth. It was not responsive to medical treatments. The patient was clinically diagnosed as PFIC type II, and no mutation on ABCB11 gene was identified. There were multiple enhancing nodules in both lobes of the liver, which were clinically diagnosed of infantile hemangioendothelioma (Fig. 1). Her jaundice progressed to have serum total bilirubin over 24 mg/dL. Coagulation profiles progressively deteriorated with frequent requirement of fresh frozen plasma transfusion. Interestingly, serum alpha-fetoprotein (AFP) level was increased to 2,740 ng/mL. The body weight of the patient was only 7 kg, thus the possibility of split LT was low within a short waiting period. Thus, we decided to perform LDLT under the clinical diagnosis of PFIC and hemangioendothelioma.
The donor was her 35-year-old mother, and the left lateral section graft weighed 216 g, being equivalent to a graft-to-recipient weight ratio of 3.1%. The graft left hepatic vein orifice was enlarged with patch venoplasty according to our institutional guidelines. Recipient operation was performed according to standard procedures of pediatric LDLT using a left lateral section graft. The native liver was cirrhotic, but there was no abnormality in the hilar vascular structures. Hepatic vein stumps of the recipient were unified to enlarge the orifice. Size-matched reconstruction of the graft hepatic vein was performed. Branch patch of the recipient portal vein was used for graft portal vein reconstruction. Single left hepatic artery reconstruction using surgical microscopy and Roux-en-Y hepaticojejunostomy were performed.
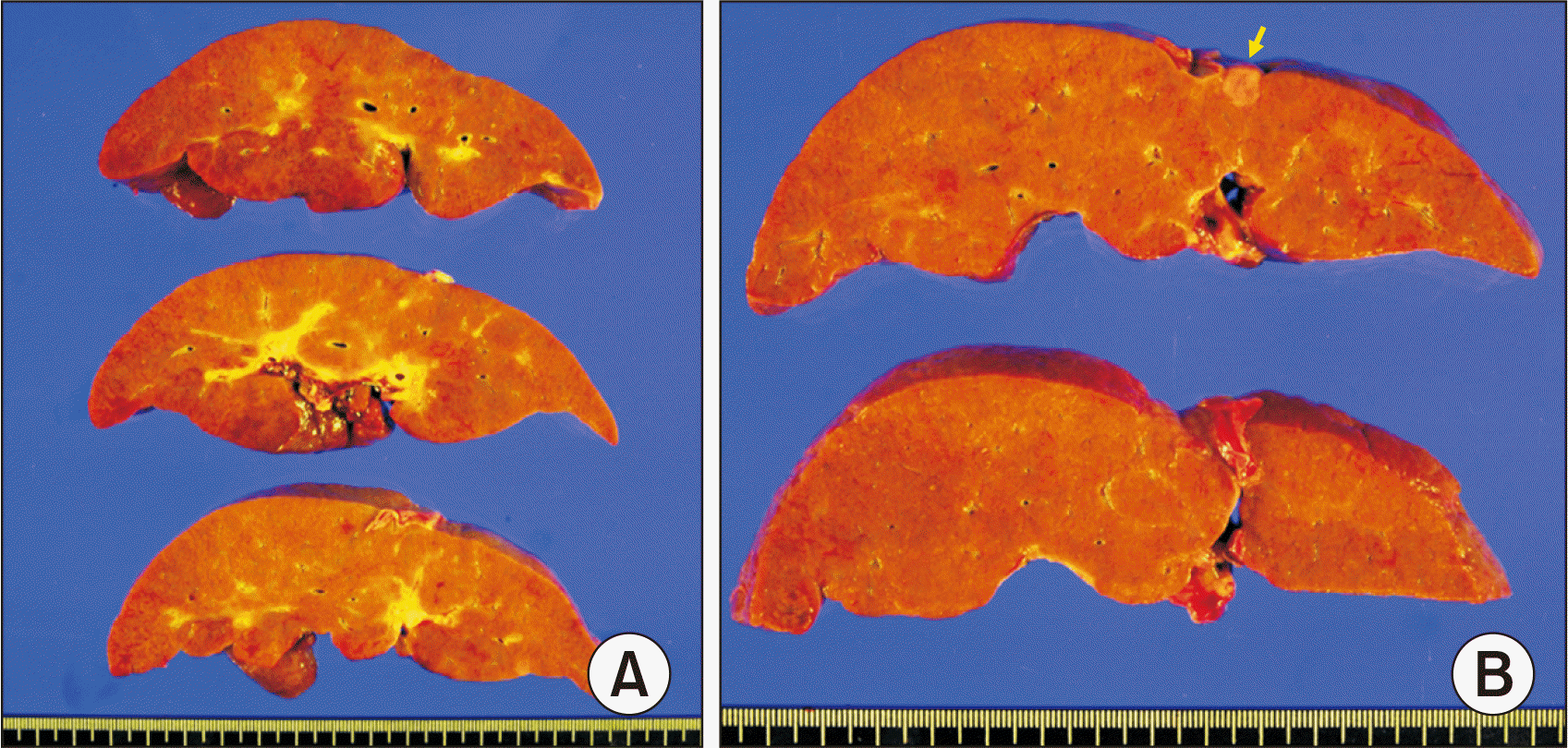
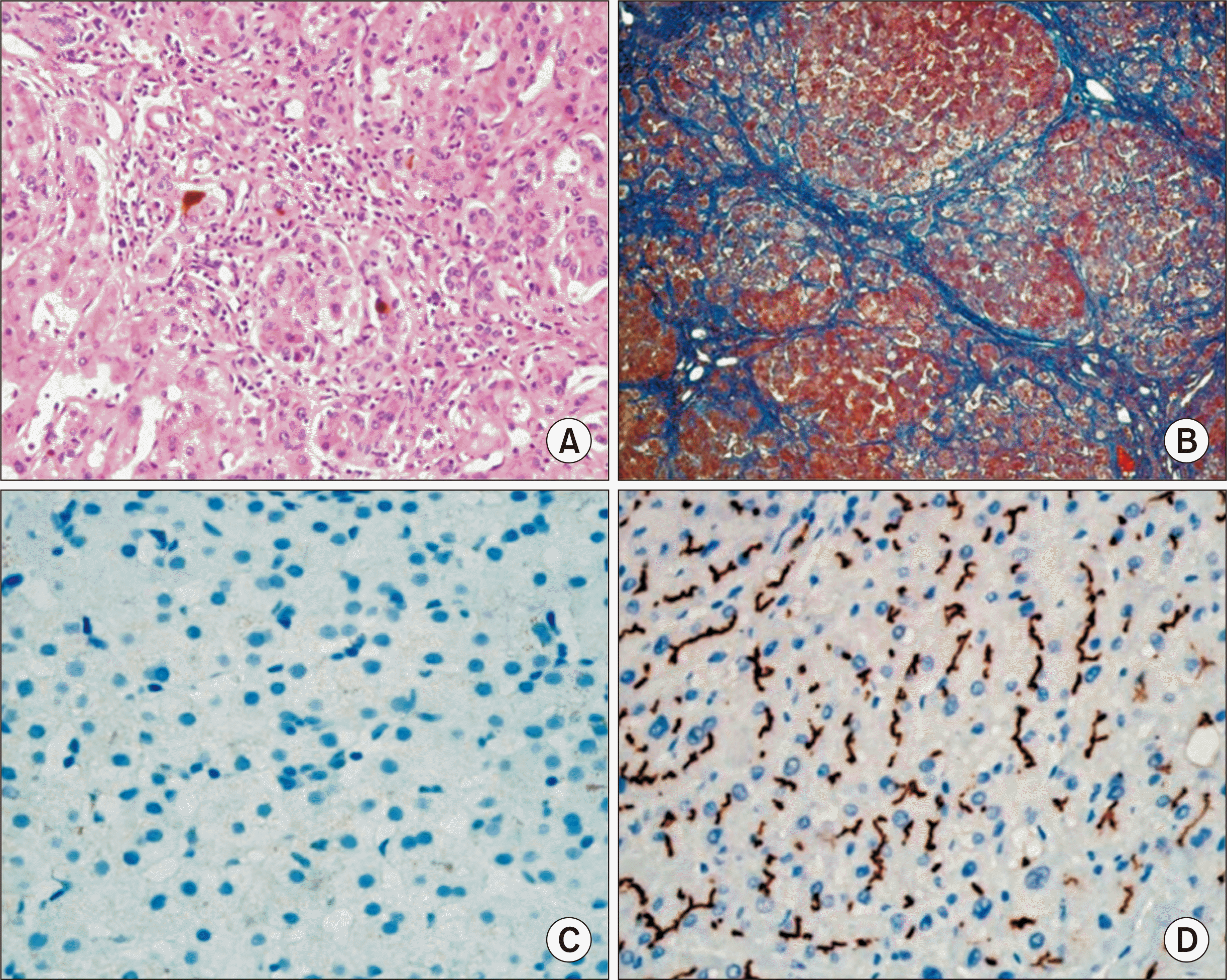
The explant liver showed micronodular liver cirrhosis, with cholestasis and bile plugs, giant cell transformation of hepatocytes, marked ductular proliferation and extramedullary hematopoiesis (Fig. 2). Overt liver cirrhosis with diffuse lobular fibrosis was identified. Immunohistochemical staining studies showed total loss of canalicular expression for bile salt export pump (BSEP) (Fig. 3). There were multiple infantile hemangiomas in both lobes of the liver with size up to 1.1 cm. In addition, there were two HCCs of 0.7 cm and 0.3 cm (Fig. 2). Tumor differentiation was Edmondson-Steiner grade II/II. Microvascular invasion, tumor necrosis, and satellite nodule were not identified.
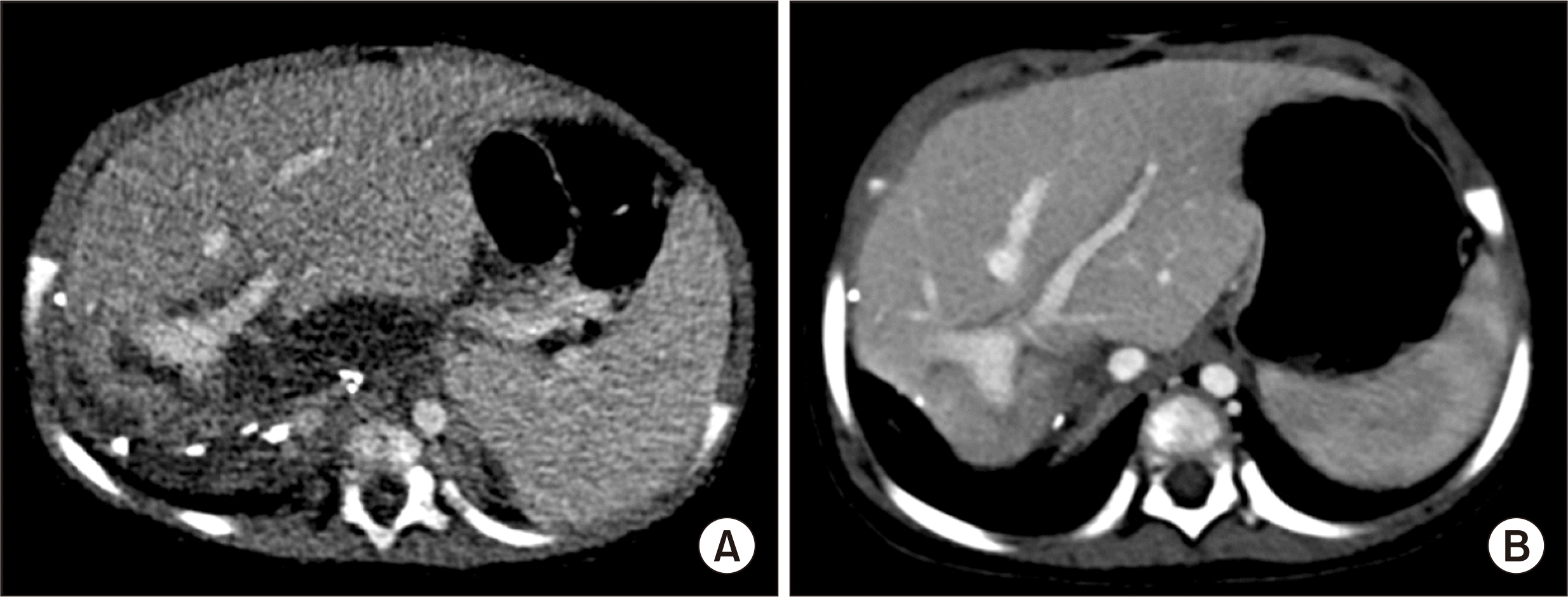
Early follow-up computed tomography scan showed no vascular complications (Fig. 4). The patient recovered uneventfully from the LDLT operation. She has been regularly followed up with abdomen ultrasonography and AFP measurement every 6 months (Fig. 5). The patient has been doing well continually for 8 years after the transplantation.
DISCUSSION
LT has been accepted as the only effective treatment for PFIC patients with end-stage liver disease. Indications for LT include severe pruritus, significant growth retardation, liver cirrhosis, and liver failure. PFIC patients usually have growth retardation and metabolic bone disease, mainly related to impaired vitamin D and calcium absorption due to reduced bile secretion. Growth retardation can be significantly improved after LT [3,4]. In the present case, the patient also has a growth curve equivalent to normal percentiles.
PFIC is classified into three types (PFIC I-III): PFIC type I is caused by a mutation of the ATP8B1 gene encoding the FIC1 protein [1], PFIC type II is caused by a mutation of ABCB11 gene encoding the BSEP. PFIC type III is due to a mutation of ABCB4 gene encoding the MDR3 protein.
Hori et al. [9,10] have reported 14 patients who underwent LDLT for PFIC (11 cases of PFIC type I and 3 cases of PFIC type II). Three of the 11 PFIC type I recipients died, while all three PFIC type II recipients survived. Eight of the 11 PFIC type I recipients developed steatosis after LDLT. Nine of these 11 PFIC type I recipients developed fibrosis after LDLT. In contrast to PFIC type I recipients, PFIC type II recipients did not develop steatosis or fibrosis after LDLT. Liu et al have reported that two cases of PFIC type I developed steatohepatitis within 1 year after LT, thus suggesting the necessity of long-term follow-up [4]. PFIC type II patients with some subtypes are prone to recurrence of liver diseases [11].
In pediatric patients, HCC is a very rare disease. Most HCCs arise in the setting of prior liver diseases [2]. They might be diagnosed incidentally in the explant liver or detected during surveillance of underlying disease. Our present case showed a high level of AFP, but the diagnosis of HCC was not taken into account, and the multiple hepatic tumors were considered as infantile hemangioendothelioma. Infantile hepatic hemangioendothelioma is the most common hepatic vascular tumor in children [12]. In a single-center experience of 28 cases, the median age at diagnosis was 3 months. Eleven patients did not receive any treatment, whereas one patient underwent resectional surgery. Sixteen patients received drug treatment, nine of whom responded well to first-line propranolol/prednisolone, whereas seven patients needed salvage treatment. Twenty-five patients are alive, whereas three patients have died [12]. In 15 adult cases of hepatic epithelioid hemangioendothelioma who underwent hepatic resection or LT at our institution, no patient showed elevated AFP levels at all [13,14].
HCC has been rarely diagnosed in infant patients with biliary atresia or other disease-associated liver cirrhosis [5-8]. We identified multiple hepatic tumors in the present case, but HCC was not considered despite high rise of serum AFP because of the extreme rarity of HCC in infants. To the best of our knowledge, there are only five cases of HCC in infant LT recipients less than 1 year of age in the literature (Table 1).
In conclusion, LT is currently the only effective treatment for PFIC-associated end-stage liver diseases. HCC can develop at the cirrhotic liver of any cause, thus elevation of serum AFP in pediatric patients is an important clue to perform further investigation before LT.
 XML Download
XML Download