

 PDF
PDF Citation
Citation Print
Print



서 론
조기영아성간질성뇌병증(early infantile epileptic encephalopathy, EIEE)은 이른 나이에 발현되는 중증의 뇌질환으로, 생후 1개월 이내에 빈번한 강직성 연축과 뇌파상 ‘suppression-burst pattern’ 소견을 보이는 Ohtahara 증후군이 대표적이나 현재는 영아성 연축(infantile spasm), Lennox-Gastaut 증후군, Dravet 증후군 등 다양한 질환을 총칭한다[1]. EIEE와 연관된 유전자는 차세대염기서열분석(next-generation sequencing, NGS) 기술이 발전함에 따라 다양하게 밝혀지고 있다[2]. 현재 EIEE와 관련하여 Online Mendelian Inheritance in Man (OMIM) database에는 약 60여개의 관련 유전자가 등재되어 있고 HCN channel 단백질을 암호화하는 HCN1 유전자도 포함되어 있다[3].
HCN1 유전자가 생성하는 HCN1 단백질은 과분극-활성화 고리형 뉴클레오티드 개폐 채널(hyperpolarization-activated cyclic-nucleotide gated channel, HCN channel) 단백질 중 포유류에서 발현되는 4가지 동형(isoform) 중 하나로, 중추 및 말초신경계에서 광범위하게 발현되고 임계 전 전위(sub-threshold potential)에서 개방되어 휴지막전위(resting membrane potential)의 안정화, 활동전위 임계(action potential threshold) 조절, 박동조율성(pacemaking) 신경활동 및 수지상세포 결합(dendritic integration) 등에 관여하는 것으로 알려져 있다[4]. 이러한 HCN1 단백질의 이상에 의해 신경생리학적 이상과 간질성 뇌질환이 유발됨이 다양한 동물 실험 및 임상 증례를 통해 보고되었다[5-10].
본 연구에서는 EIEE로 진단받은 환자에서 NGS 검사 및 부모의 Sanger sequencing 검사를 통해 기존에 보고되지 않았던 HCN1의 de novo 돌연변이를 검출하였던 사례를 소개하고자 한다.
증 례
특별한 가족력, 과거력이 없는 여아가 생후 4개월 때부터 전신 경련성(generalized clonic) 발작이 지속되어 phenytoin, keppra, trileptal, sentil 등의 약물치료를 받았으나 증세가 호전되지 않아 생후 7개월 때 본원으로 전원되었다. 발작의 양상은 초기에 전신 경련성 발작을 보이다 생후 6개월 때부터 상지 경련성 운동(arm clonic movement)을 동반한 무호흡성 경련(apnea seizure)으로 바뀌었으며, 열성 경련(febrile seizure)과 비열성 경련(afebrile seizure)이 혼재돼 있었다. 본원으로 전원될 당시 체온은 37.8°C로 미열이 간헐적으로 관찰되었고 뇌파 검사상 ‘mild slow & disorganized background rhythm’ 소견이 관찰되어 경도의 미만성 뇌병증(diffuse encephalopathy) 소견을 보였다.
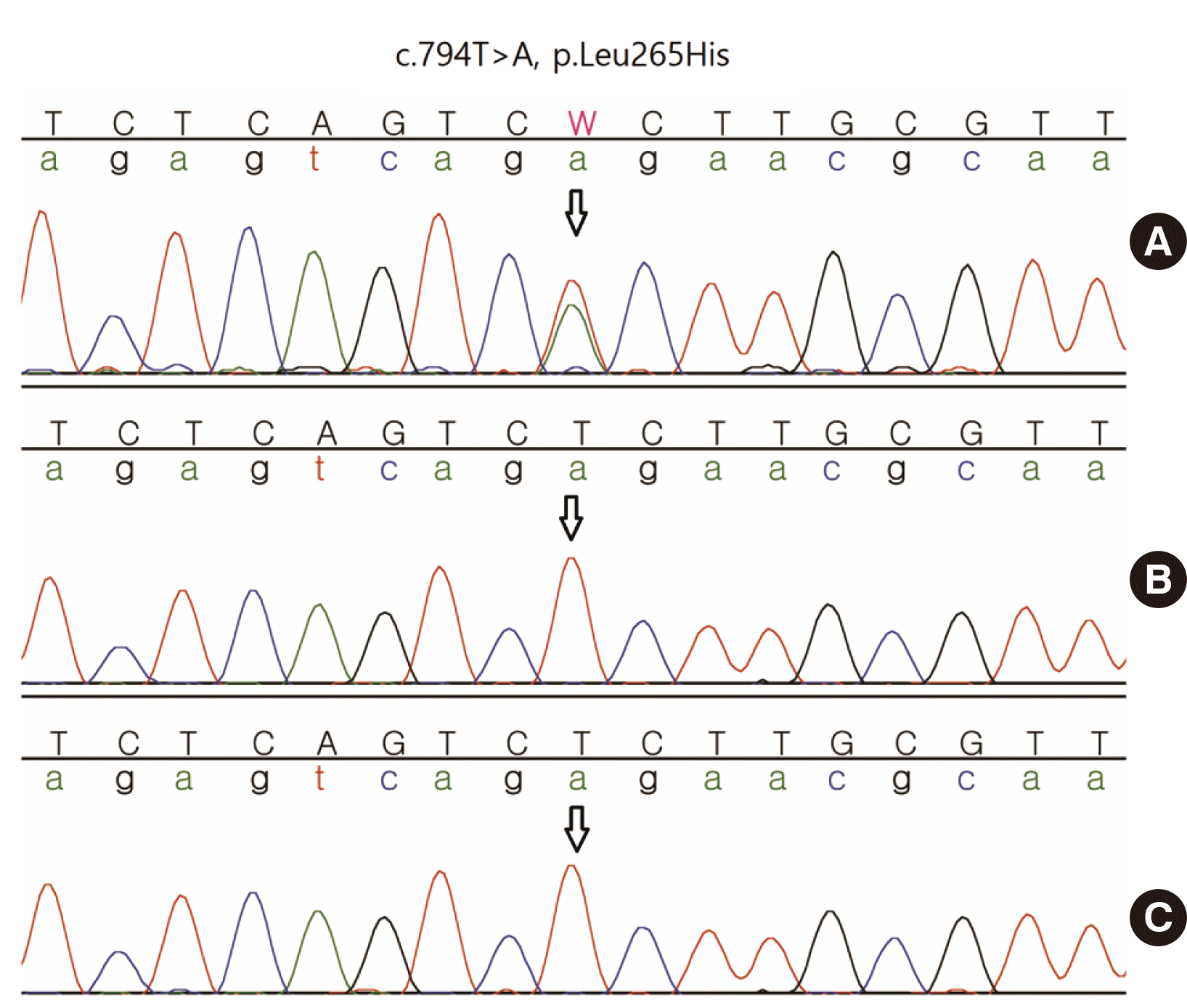
환아의 간질성 뇌병증에 대한 선천적인 원인을 파악하기 위해 환아의 말초혈액 검체를 이용하여 NGS를 기반으로 한 신경발달 유전자 패널(neurodevelopmental panel) 검사를 진행하였다(NextSeq 550Dx System; Illumina, San Diego, CA, USA). 해당 패널에는 HCN1을 포함하여 신경발달 질환과 연관된 약 4,870개의 유전자가 포함되어 있다. 환아의 NGS 검사 결과 염색체나 유전자의 복제수 변이는 발견되지 않았고, 12개의 VOUS (variant of unknown significance)로 해석되는 단일염기 변이가 발견되었다(Table 1). 이 중 HCN1 유전자의 c.794T>A, p.Leu265His (accession #: NM_021072.3) 변이는 환아의 임상 증상과 연관 가능성이 높았고 gnomAD/ExAC (https://gnomad.broadinstitute.org/), 1000 Genomes Project (http://1000genomes.org), Korean Reference Genome Database (KRGDB, http://coda.nih.go.kr/coda/KRGDB/index.jsp) 등의 데이터베이스에서도 빈도가 검색되지 않는 희귀 변이였다(PM2). 또한 웹 기반 in silico analysis상 SIFT (score 0)/MutationTaster (score 1)/FATH MM (score 5.1)/MetaSVM (score 1.103)/REVEL (score 0.972) 등 21개의 분석 알고리즘상 질병연관 변이일 가능성이 높을 것으로 예측되었다(PP3). 하지만 본 변이는 질환과 관련하여 이전 문헌에 보고된 적이 없었기 때문에 특이 과거력이 없었던 부모를 대상으로 Sanger sequencing 검사 및 생물학적 부모 여부 확인을 위해 16개의 마커를 이용한 STR (short tandem repeat) 검사를 추가로 진행하였다. 검사 결과 친자 관계를 확인할 수 있었으며, 부모 모두 환아에서 발견된 HCN1 유전자의 돌연변이가 관찰되지 않았기 때문에(Fig. 1) de novo임이 확인되었다(PS2). 2015 American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines에 따르면, 1개의 strong evidence (PS1-PS4)와 1-2개의 moderate evidence (PM1-PM6)가 동시에 부여되는 것 이상이면 병적인 가능성이 있는(likely pathogenic) 변이로 판정할 수 있다[11]. 상기 기준에 근거하여 1개의 친자 및 de novo 확인과 관련된 strong evidence (PS2)와 1개의 정상집단에서 발견되지 않는 변이와 관련된 moderate evidence (PM2) 및 1개의 in silico algorithms 결과와 관련된 supporting evidence (PP3)를 조합하면, 상기 변이는 병적일 가능성이 있는(likely pathogenic) 변이로 예측될 수 있다. HCN1 유전자 이외에 단일염기 변이가 관찰된 유전자 중 TBC1D24, GFPT1, TMEM67, PMM2, AP4M1, SLC45A1, RPGRIP1L, APC2 유전자는 상염색체 열성 유전자인데 환자에서는 각각 한 개의 변이만 확인되었고, NOTCH3와 GLUD1 유전자는 환자의 임상 증상과 연관성이 낮았다. 또한 KCNMA1 유전자의 변이는 정상 인구집단 빈도가 높았다(ExAC Global: 0.83%, KRGDB: 4.42%). 이러한 이유로 HCN1 유전자 변이 이외에 다른 유전자 변이들에 대해서는 추가 부모검사 없이 모두 VOUS (variant of unknown significance)로 해석하였다(Table 1). 환아는 본원으로 전원 후 일주일간 입원치료를 받았으며 입원기간 동안 trileptal, sentil, sabril, phenytoin, keppra 약제를 투여받아 발작이 조절되었다. 퇴원 시 phenytoin, keppra 약제를 중단하였고 외래에서 trileptal, sentil, sabril을 투약하며 현재 경과 관찰 중이다.
고 찰
HCN channel은 신경세포의 흥분도를 음성 피드백 루프를 통해 안정화시키는 역할을 담당한다[12]. HCN1 단백질은 중추신경계에서 주로 발현되는 동형(isoform) 중의 하나로[5, 6], HCN1의 하향조절(down-regulation) 또는 기능 저하에 의해 발작(seizure) 증상의 중증도가 심해지고 그에 의한 사망률이 높아짐이 mouse knockout 실험을 통해 알려져 뇌전증과의 관련 가능성이 보고되었다[8]. 이후 EIEE 환자들에게서 발견된 HCN1 변이에 대한 연구가 이루어져, HCN1 단백의 상향조절(upregulation)이 유발되는 경우에도 특발성 전신성 뇌전증(idiopathic generalized epilepsy)이 발생할 수 있음이 보고되었다[9]. 해당 연구에서 HCN1 변이를 지닌 환자들은 공통적으로 4개월에서 13개월 사이에 시작되는 열성/비열성 혼합형(polymorphic) 발작을 보이다가 Dravet 증후군과 다르게 근경련성 수축(myoclonic jerk)이 동반된 비특이적 결신 발작(atypical absence seizure)을 보이며 국소적 발작(focal seizure)으로 진행하는 양상을 보였다고 한다. 상기 양상은 본 증례에서 보인 증상의 발현 시기(생후 4개월) 및 경련의 초기 양상(열성/비열성 혼합형)과 유사했고, 이후 결신 발작 혹은 국소적 발작으로 진행하는지 추적관찰이 필요하다.
HCN channel 단백은 공통적으로 6개의 막관통 단위(transmembrane segment)인 S1–S6 단위와 C-terminal 부위의 고리형 뉴클레오티드 결합 부위(cyclic nucleotide-binding domain)로 이루어진 구조를 공유하며 S5, S6 단위 사이에 존재하는 동공 루프(pore loop) 구조를 통해 이온을 통과시키는 구조이다[13]. 본 환자에서 발견된 HCN1 유전자의 c.794T>A, p.Leu265His 변이는 HCN1 단백질의 S4 단위상에 존재한다. S4 단위는 양전하 아미노산(positive-charged amino acid)들을 다수 포함하고 있어 세포막의 전기장 변화에 반응한다[14]. EIEE 증례에서 발견된 HCN1 유전자의 de novo missense 변이들에 대해 기능적 연구(functional study)가 있었고, S4 단위와 연관된 돌연변이 중 His279Tyr의 경우 기능향상변이(gain-of-function variant)로 작용하는 반면 Ser272Pro와 Arg297-Thr의 경우 야생형(wild type) 단백에 대해 우성저하효과(dominant-negative effect)를 보였다[9]. 본 증례에서 발견된 Leu265His 변이는 비극성(non-polar) 아미노산이 양전하 아미노산으로 대체되는 missense 변이로, HCN1 단백이 세포막의 전기장 변화에 반응하는 민감도를 향상시킬 것으로 예측되나 상기 연구에서와 같이 missense 변이의 영향은 다양한 양상을 보일 수 있으므로 정확한 기능 변화를 판정하기 위해서는 추가적인 연구가 필요하다.
HCN channel은 신경 흥분도 조절과 관련된 약제의 표적으로서 주목받고 있다[15]. 특히 HCN channel이 발현되는 조직은 심장과 신경계에 국한되어 있기 때문에 HCN channel 표적치료제는 호흡 작용 및 혈관 평활근 긴장도(vascular smooth-muscle tone)에 대한 부작용 없이 심장질환 및 신경계질환에 치료효과를 보일 것으로 기대된다. 뇌전증과 HCN channel 기능 간의 관계는 복잡하고 다양하여 항뇌전증제의 경우 HCN channel에 대한 약리학적 차단 및 활성이 모두 경우에 따라 긍정적인 영향을 미치는 것으로 알려져 있다[12]. Gabapentin, lamotrigine 등 기존에 알려진 항뇌전증제의 경우 HCN channel 기능을 상향 조절하는 효과가 있음이 알려졌으며[16, 17], 이와 반대로 HCN channel 억제제가 뇌전증 치료효과를 보일 수 있음이 보고되기도 하였다[18]. HCN1의 기능획득변이에 의한 질환의 경우 HCN1의 공동 구조에 결합하는 물질이 억제제로 사용될 수 있으나, 기존의 약제들은 단백 친화도와 특이도가 낮기 때문에 새로운 억제제 후보물질에 대한 연구 또한 활발히 진행되고 있다[4, 15]. 본 증례에서 발견된 Leu265His 변이에 대해 기능적 연구가 추가적으로 시행된다면 향후 맞춤형 표적치료제를 이용한 치료계획 수립에 기여할 수 있으리라 기대된다.
본 증례는 기존에 보고된 바 없는 HCN1의 병적일 가능성이 있는(likely pathogenic) 변이를 최초로 검출하였다는데 의미가 있다. 또한 환자의 예후 및 치료계획 수립 시 유전자 검사의 중요성에 대한 경험을 공유할 수 있어 간질 혹은 신경발달 장애 관련 NGS 검사를 시행하는 국내외 다른 임상검사실 및 임상의사에게 도움이 될 것으로 생각된다.
 XML Download
XML Download