

 PDF
PDF Citation
Citation Print
Print



가족성 고콜레스테롤혈증은 유병률이 약 1:250으로 알려진 흔한 유전 질환으로[1], 저밀도지단백 콜레스테롤이 지속적으로 상승되어 있는 이상지질혈증이다. 이로 말미암아 죽상경화성 플라크가 관상동맥을 비롯한 여러 혈관에 축적되고 혈관이 좁아지면서 조기 심혈관계 질환 발병의 주요 위험 인자로 작용한다. 관상동맥질환으로 인한 사망률은 우리나라뿐 아니라 전세계적으로 증가하고 있으므로[2] 적절한 치료, 나아가 이른 진단은 반드시 필요하다고 할 수 있으나 이런 중요성에도 불구하고 가족성 고콜레스테롤혈증은 과소 진단 및 과소 치료가 이루어지고 있다고 보고된 바 있다[3, 4].
가족성 고콜레스테롤혈증의 대부분은 LDLR, APOB, PCSK9 등 세 유전자의 이형접합 병인성 변이로 발생한다고 알려져 있으며, 이들 중에서도 저밀도지단백 수용체를 코딩하는 LDLR 유전자의 변이가 대부분을 차지한다[5]. 현재까지도 대표적으로 사용되고 있는 가족성 고콜레스테롤혈증의 진단 기준(criteria)들에는 네덜란드의 Dutch Lipid Clinic Network diagnostic criteria [6], 영국의 Simon Broome diagnostic criteria for Familial Hypercholesterolemia [7]가 있으며 앞서 언급한 세 유전자의 변이가 존재할 경우 가족성 고콜레스테롤혈증 진단에 있어 높은 점수를 부여하고 있다.
LDLR 유전자의 변이는 90% 이상이 염기서열분석법(sequencing)으로 확인이 가능하나, 일부는 다중결찰의존탐사체증폭(multiplex ligation dependent probe amplifi-cation) 방법으로 확인이 가능하다고 알려져 있다[8]. 한편, 유전 분석 기술의 발달로 차세대염기서열분석법(next-generation sequencing)과 같은 검사법이 등장하여 단일 유전자가 아닌 여러 유전자들을 패널 단위로 한꺼번에 분석하는 것도 가능해졌다.
유전 분석에서 확인된 변이들은 미국의학유전학회(American College of Medical Genetics, ACMG)와 분자병리학회(Association for Molecular Pathology, AMP)에서 제시한 변이의 해석에 대한 종합적 권고지침(이하 ACMG/AMP 지침)에 따라 pathogenic variant (PV), likely pathogenic variant (LPV), variant of uncertain significance (VUS, 미분류염기변이), likely benign variant (LBV), benign variant (BV) 등의 다섯 범주 중 하나로 분류된다[9]. 그러나 ACMG/AMP 지침은 개별 유전자 또는 질환의 고유한 특징을 고려하지 않았기 때문에, 질환별 전문가 집단(specific disease group)별로 해당 질환에 더욱 초점을 맞춘 가이드라인을 개발할 것을 권고하고 있다[9].
2013년 Clinical Genome Resource (ClinGen) 컨소시엄[10]이 설립된 이후, ClinGen 산하의 Familial Hypercholesterolemia Variant Curation Expert Panel (FH-VCEP) 중 일부 구성원들이 LDLR 유전자의 특성을 고려한 ACMG/AMP 지침의 적용을 제안한 바 있다[11]. ClinGen FH-VCEP에서 2020년 9월 정식 가이드라인 1판을 발행한 이후, 2021년 4월 ClinGen FH-VCEP Specifications to the ACMG/AMP Variant Classification Guidelines Version 1.1 (이하 Clin-Gen 가이드라인)을 발행하였고, LDLR 유전자의 변이 해석에 사용하도록 명시하였다[12]. 기존의 ACMG/AMP 지침에서 변화된 내용으로는 기능 연구(functional study)의 변이 부여(PS3, BS3) 기준이 구체화되고 세분화되었으며, PM2, BA1, BS1 등의 기준이 되는 대립유전자빈도(minor allele frequency)가 구체화된 점 등이 있다.
한편, 앞서 기술한 ACMG/AMP 지침의 다섯 범주 중 VUS로 분류된 변이들의 경우에는 해석, 보고 여부, 결과 설명, 재분석 등의 관점에서 복잡성이 존재하는데[13], LDLR 유전자의 VUS를 평가하거나 재분석한 국내 보고는 없었다. 이에, 저자들은 기존에 시행한 LDLR 유전자 변이 분석에서 VUS로 보고된 변이들을 중심으로 ClinGen 가이드라인을 적용해 재분석하여 ACMG/AMP 지침[9]의 다른 범주로 재분류할 수 있는지 확인하고자 하였다.
이 연구는 본원의 임상시험심사위원회의 승인(승인번호: 2021-05-065)을 받아 진행하였다. 먼저, EDTA 용기에 얻어진 환자의 말초 혈액을 Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA)를 이용하여 제조사에서 제시한 방법으로 genomic DNA를 얻었다. LDLR 유전자(NCBI 표준염기서열 NM_000527.5)의 염기서열분석을 위해 18개의 코딩 엑손 모두와 각각의 인접 인트론 부위(25 bp 이내)를 PCR을 시행하였다. PCR은 GeneAmp PCR system 9700 thermal cycler (Applied Biosystems, Foster City, California, USA)를 이용하였으며, 직접염기서열분석은 BigDye Terminator Cycle Sequencing Ready Reaction Kits (Applied Biosystems)와 ABI 3130xl genetic analyzer (Applied Biosystems)를 이용하였다. 확인된 LDLR 유전자의 변이들은 Human Genome Variation Society (HGVS, http://varnomen.hgvs.org/)의 명명법[14]에 따라 기술하였고, 코딩 DNA 표준 염기서열에서 번역의 개시 코돈 ATG의 첫번째 뉴클레오티드 “A”를 “c.1”로 정의하였다. 확인된 변이들은 ACMG/AMP 지침에 따라 PV, LPV, VUS, LBV, BV 중 하나의 범주로 분류하였다.
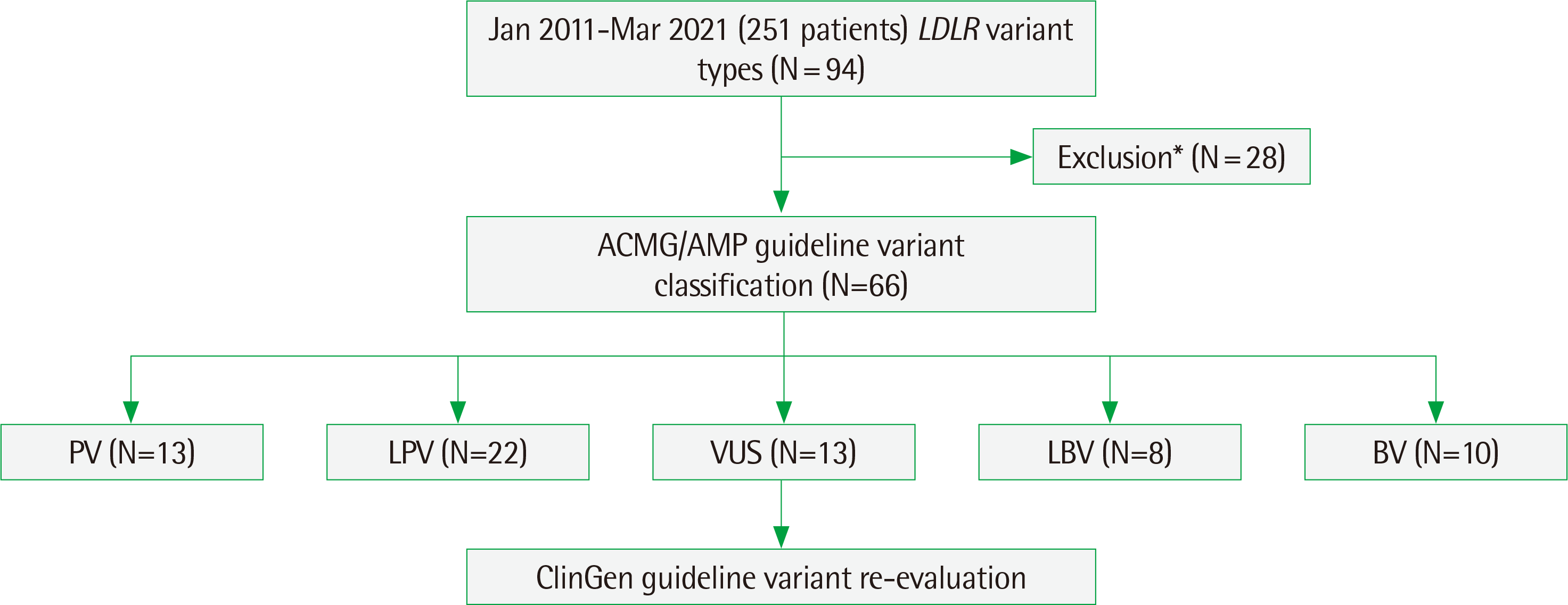
저자들은 2011년 1월부터 2021년 3월까지 LDLR 유전자 검사가 의뢰되어 결과가 보고된 251건 가운데 15명의 환자에서 VUS로 보고한 서로 다른 13종류의 변이들을 ClinGen 가이드라인[12]에 따라 재판독하였다[9](Fig. 1). 본원에서 VUS로 보고한 변이들이 다른 연구들에서도 보고된 적이 있는지를 확인하기 위하여 The Human Genome Mutation Database (HGMD® Professional release 2021.1, Institute of Medical Genetics, Cardiff, Wales; https://my.qiagendigitalinsights.com/bbp/view/hgmd/pro/start.php)와 ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/) 데이터베이스를 이용하였으며, 앞의 데이터베이스에서 동일 코돈에서 다른 아미노산으로의 변화를 일으키는 변이가 보고 되었는지도 확인하였다. 재분류를 시행한 변이들의 인구집단별 빈도는 gnomAD v2.1.1 (https://gnomad.broadinstitute.org/) 데이터베이스를 이용하여 조사하였다. 과오 변이(missense variant)들의 경우in-silico 분석을 위한 REVEL score [15] 조사는 ClinGen (https://curation-test.clinicalgenome.org/select-variant/)을 이용하였으며, 동의 변이(synonymous variant)나 비전형적인 인트론 위치(non-canonical splice acceptor and donor sites) 또는 엑손-인트론 경계(3 bp 이내)에서 확인된 변이들의 경우 짜깁기 영향(splicing effect)을 확인하기 위한MaxEntScan (MES) score [16] 조사는 Alamut Visual v.2.15 (SOPHiA GENETICS, Saint-Sulpice, Switzerland) 프로그램을 이용하였다. 저밀도지단백 콜레스테롤 및 연관검사결과, 신체 진찰 소견, 관상동맥질환과 고콜레스테롤혈증의 가족력 등은 의무기록을 통해 확인하였고, 외부의료기관으로부터 검사가 의뢰된 경우에는 검사의뢰지에 기재된 관련 임상정보 등을 판독에 활용하였다.
또한, 본원에서 LPV 또는 LBV로 보고한 변이들을 VUS와 마찬가지로 ClinGen 가이드라인에 따라 재분류하여 VUS로 변경되는 변이가 있는지 확인하였다.
재분석 대상이었던 13종류의 VUS를 유형별로 보면, 과오 변이가 8종류(61.5%)였으며, 인트론 위치의 변이가 4종류(30.8%), 동의 변이가 1종류(7.7%)였다. 13종류의 변이들 중 HGMD에 기존 보고가 있었던 변이는 5종류(38.5%)였으며, Clinvar에 보고된 바 있는 변이는 10종류(76.9%)였고 그 중 3종류의 변이는 Clinvar에서 여러 보고자들 간의 변이 해석이 서로 일치하지 않았다.
재분석 결과 13종류의 변이들 중 2종류(15.4%)의 변이가 LPV로 범주가 변경되었는데, c.268G>T (p.D90Y) 변이(PM2, PM5_Strong, PP3)와 c.694G>T (p.A232S) 변이(PS3_Supporting, PM1, PM2, PP3)가 이에 해당하였다. 나머지 11종류(84.6%)는 VUS로 유지되었고 재분류 결과 PV, LBV 또는 BV로 조정된 변이는 없었다(Table 1). 또한, 8종류의 LBV 중 c.344G>A (p.R115H) 1개 변이가 VUS로 재분류되었고(12.5%), 22종류의 LPV 중 c.1228A>G (p.Arg410Gly)와 c.1571_1573del (p.Val524del) 등 2개 변이가 VUS로 재분류되었다(9.1%) (Table 1).
가족성 고콜레스테롤혈증의 높은 유병률[1]과 유전적 원인[5]을 고려하면 LDLR 유전자의 변이들은 매우 많이 존재할 수 있으며, Clinvar 데이터베이스의 가족성 고콜레스테롤혈증 연관 유전자들의 변이를 분석한 최근 논문에 따르면 2,314개의 서로 다른 LDLR 유전자 변이가 존재하였다[17]. 해당 문헌에 따르면 “Uncertain significance”로 분류된 변이는 182종류(7.9%)였으며 “Con°icting classification”으로 분류된 변이는 303종류(13.2%)였다[17]. 다른 공용 데이터베이스에서 얻은 LDLR 유전자 변이 1,894개를 분석한 연구에서는 795종류(42.0%)의 변이가 VUS로 분류되었다[11]. 후자의 연구에서는 대립유전자형 빈도가 5%를 초과한(ACMG/AMP 지침[9]의 BV에 해당하는) 변이들이 분석에서 제외되었음을 고려하여도, VUS 변이들이 높은 비율로 존재하고 있음을 알 수 있다.
특정 변이가 (L)PV나 (L)BV와 같은 변이로 분류되지 못하고 VUS로 보고하게 되는 요인으로는 가족 내 분리(segregation) 현상(PP1 부여), 표현형(PP4), 기능 연구(functional study, PS3) 등의 정보가 불충분하여 변이에 대한 근거를 부여하지 못하는 점이 제시된 바 있다[11, 17]. 본원에서 시행한 LDLR 유전자검사 중 대부분(225건, 89.6%)이 외부의료기관으로부터 의뢰된 경우였는데, ACMG에서도 유전자검사 결과의 재분석에 있어 환자의 표현형과 가족력 등을 언급하고 있다는 점[18]을 고려하면, 의뢰서나 의무기록에서 가족력, 신체 진찰 소견, 또는 연관검사결과 등의 임상 정보가 충분히 제시된다면 PP1, PP4 등의 근거를 추가로 부여하여 VUS가 LPV나 다른 범주로 재분류 될 가능성이 있다고 판단된다. 기존 문헌에 보고된 적이 없는 c.781T>A (p.C261S) 변이의 경우, supporting evidence가 하나 모자라 ClinGen 가이드라인을 이용하여 재판독을 하였음에도 불구하고 변이 분류에서 VUS가 유지되었다(Table 1). ClinGen 가이드라인을 활용하여 변이를 해석할 때에는 환자의 저밀도지단백 콜레스테롤, 가족력, 신체 진찰 소견 등이 충분히 제시되어 Dutch Lipid Clinic Network 점수가 6점 이상이라면 PP4의 추가 부여가 가능해지므로 LPV로 재분류될 수 있다.
기존의 분류체계에서 VUS였다가 LPV로 재분류된 변이들의 경우, ACMG/AMP 지침의 근거에 PM1을 추가로 부여할 수 있었거나, PM5 대신 PM5_Strong을 부여할 수 있는 특징이 있었다. 이 두 근거를 분석해 보면, 변이가 발생한 코돈의 중요성을 FH-VCEP에서 ClinGen 가이드라인에 반영한 결과로 생각한다. 기존의 ACMG/AMP 지침에서는 PM1을 부여하는 기준에 대해 세부적으로 명시하지 않았으며, 본원에서는 영국의 Association for Clinical Geno-mic Science (ACGS)에서 제시한 ACGS 2020 가이드라인[19]에서 명시한 FBN1 유전자의 EGF-like calcium-binding domain 내의 cysteine, NOTCH3 유전자의 EGF-like repeat 내의 cysteine, (COL1A1 등의) 콜라겐을 인코딩하는 유전자들에서 glycine의 과오 변이와 같은 제한된 경우에만 PM1을 사용해 왔다. 하지만 이번 ClinGen 가이드라인을 통해서 moderate evidence를 하나 더 줄 수 있는 근거가 세부적으로 마련된 의미가 있다.
PM5_Strong을 부여하여 LPV로 재분류된 변이(c.268G>T, p.D90Y)의 경우, 해당 코돈이 HGMD에서 p.D90A, p.D90N, p.D90E, p.D90G와 같은 (L)PV로 보고된 바 있다. 이 변이는 두 가지 관점에서 의미를 갖는다. 첫째, 해당 코돈에서 네 가지의 서로 다른 아미노산으로의 변화가 보고되었다는 것은 고보존되는(highly conserved) 중요 영역임을 시사한다. 그럼에도 기존에는 이 변이에 PM5만 부여할 수 있었는데, ClinGen 가이드라인을 적용한 이후에는 해당 코돈의 중요성에 대한 가중치가 반영되어 근거의 강도를 높일 수 있었다. 둘째, 변이가 발생한 코돈이 ClinGen 가이드라인에서 제시하는 엑손 4의 과오 변이 또는 cysteine의 과오 변이가 아니라 할지라도, 그 위치가 중요한 부위라면 병인성 변이를 시사하는 근거(본 변이의 경우 PM5_Strong)를 부여할 수 있다는 점이다. 그러면서도 ClinGen 가이드라인에서는 PS1 또는 PM5와 PM1을 동시에 부여하지 못하게 규정함으로써 중복 계산을 방지하였고 변이 해석의 정확성을 높였다. 일례로 본원에서 LPV로 보고한 c.361T>G (p.C121G)의 경우, 엑손 4에 위치한 cysteine의 과오 변이였지만, PM1과 PM5_Strong (p.C121R, p.C121F, p.C121S, p.C121Y이 보고된 바 있음)을 동시에 부여하지 않아 기존의 분류가 유지되었다.
한편, VUS에서 LPV로 재분류된 또다른 변이인 c.694G>T (p.A232S)의 경우, ClinGen 가이드라인에서 제시한 기능 연구의 변이 부여 기준이 구체화되고 PS3, PS3_Moderate, PS3_Supporting과 같이 세분화된 점을 적용하였다. Rodríguez-Jiménez 등[20]의 연구에 따르면 본 변이는 LDL 흡수(uptake)와 LDL 결합(binding)에 대한 활성도가 야생형(wild type)과 비교했을 때 각각 73%와 79%로, ClinGen 가이드라인의 PS3_Supporting 부여 기준인 85% 미만을 충족하였다.
LDLR 유전자는 대부분(80.6%)의 변이가 세계적으로 광범위하게 확인되기보다는 유럽, 아시아, 아메리카, 아프리카, 또는 오세아니아 중 하나의 대륙에서만 집중적으로 발견된다는 점에서 변이 해석이 중요한 의미를 갖는다[11]. 대만과 중국에선 c.986G>A (p.C329Y)와 c.1747C>T (p.H583Y), 일본에선 c.1845+2T>C (p.?)와 c.1012T>A (p.C338S), 우리나라에선 c.682G>T (p.E228*)와 c.2054C>T (p.P685L) 변이가 흔한데, 이와 같이 아시아 내에서도 국가별로 흔하게 발견되는 변이가 서로 차이가 있다는 점에서 중요하다고 볼 수 있다[21]. 또한, 해마다 LDLR 유전자 검사 건수가 증가함에 따라, 새로운 변이의 발견 또한 빈번해질 것으로 생각되며 그만큼 정확한 변이 해석의 중요성 또한 부각될 것으로 생각된다.
요약하면, ClinGen 가이드라인을 이용하여 VUS 변이를 재분석한 결과 기존의 ACMG/AMP 지침[9]의 분류체계에서는 LPV로 생각되었으나 근거가 부족했던 과오 변이들을 적절히 재분류할 수 있었다. 가족성 고콜레스테롤혈증의 유병률과 LDLR 유전자가 90% 이상의 원인임을 고려하면 향후 변이 해석 시 ClinGen 가이드라인을 적극적으로 활용할 필요성이 있다.
 XML Download
XML Download