

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Adult-onset Still’s disease (AOSD), first described in 1971, is a rare inflammatory disorder of unknown etiology. It is characterized by a constellation of symptoms and signs including sore throat, daily spiking fever, arthritis, typical salmon-color rash, lymphadenopathy/splenomegaly, leukocytosis, liver dysfunction, and elevated inflammatory markers [1]. AOSD and systemic idiopathic arthritis (sJIA) have been considered as a continuum of autoinflammatory disease and are classified based on the age of onset.
Macrophage activation syndrome (MAS) is a fatal AOSD complication presenting with high fever, rash, pancytopenia, hepatosplenomegaly, hepatic dysfunction, encephalopathy, coagulation abnormality, and increased levels of ferritins. It is caused by an excessive activation of T lymphocytes and hemophagocytic macrophages and massive pro-inflammatory cytokinemia including interleukin-1β (IL-1β) and interleukin-6 (IL-6) [1].
Through advances in understanding the mechanisms of AOSD/sJIA and MAS, anti-cytokine treatments, especially IL-1β and IL-6 blockade, have been introduced for treatment-refractory cases [1]. However, the development of MAS has been rarely reported in patients with AOSD/sJIA during treatment with IL-1β or IL-6 blocking agents [2-8]. To the best of our knowledge, there have been no reports of MAS developed in AOSD patients receiving biologic therapy in Korea. Therefore, we report the first MAS case diagnosed during tocilizumab treatment.
CASE REPORT
A 20-year-old female was admitted because of an intermittent fever lasting for 1 week. She had been healthy, except for active atopic dermatitis, and was diagnosed with AOSD 34 months prior to admission (PTA). AOSD was diagnosed based on an unexplained spiking fever for 2 months, pruritic erythematous maculopapular rashes and urticarial lesions, knee arthritis, cervical and intra-abdominal lymphadenopathy, leukocytosis (white blood cells [WBC] 33,780/µL), hypertransaminasemia, elevated acute phase reactants (erythrocyte sedimentation rate [ESR] 42 mm/h; C-reactive protein [CRP] 25.2 mg/dL), hyperferritinemia (732 ng/mL), and negative antinuclear antibody and rheumatoid factor; these findings satisfied the Yamaguchi criteria [9]. Despite that methotrexate and cyclosporine were added as steroid-sparing agents, the minimal required dose of prednisolone was ≥0.5 mg/kg, and 25 months PTA, she started intravenous tocilizumab (8 mg/kg) every 4-week. After a 12-month treatment of tocilizumab with methotrexate, prednisolone was completely tapered off. Fifteen months PTA, the infusion intervals were prolonged to 6 weeks. Urticaria recurred during prednisolone tapering, and it was not related to tocilizumab. A combination of antihistamines, montelukast, and cyclosporine was not helpful for the idiopathic urticaria. Five months PTA, she tried monthly omalizumab, a humanized monoclonal anti-IgE antibody. Three weeks after the 4th injection of omalizumab and 5 weeks after the 24th tocilizumab infusion, she re-developed fever without skin rash or arthralgia. The scheduled administration of tocilizumab resulted in only a transient defervescence.
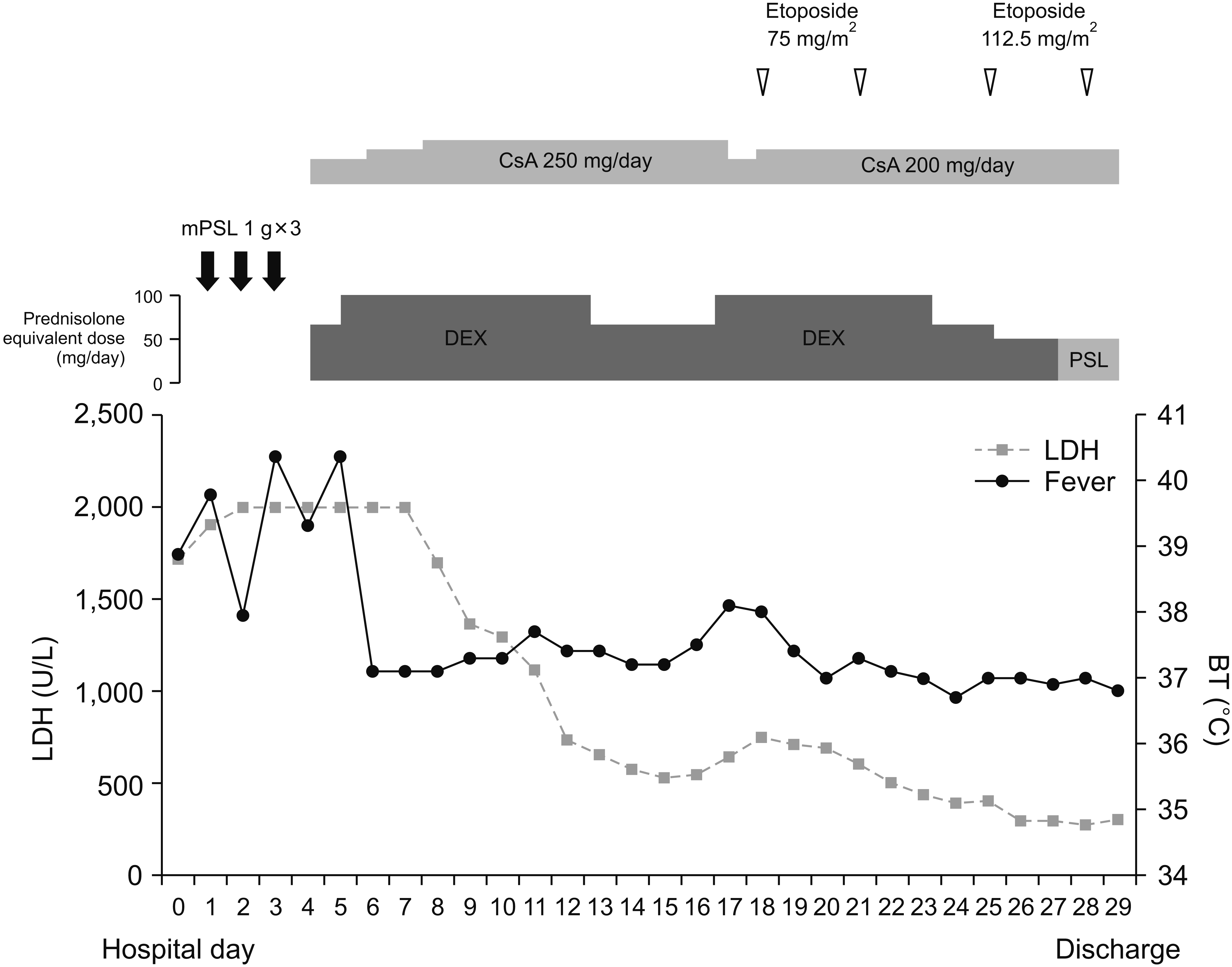
A physical examination revealed a body temperature of 39.5°C, non-tender small-sized lymph nodes in the neck and tenderness in the right upper quadrant of the abdomen. Complete blood cell counts showed a WBC count of 4,190/µL (neutrophil 86.1%, lymphocyte 7.9%) and a hemoglobin level of 12.2 g/dL. The ESR and CRP levels were 2 mm/h and <0.4 mg/dL, respectively. However, she had mild thrombocytopenia (73×103/µL), hypertransaminasemia (aspartate aminotransferase 794 IU/L; alanine aminotransferase 419 IU/L) with elevated gamma-glutamyl transferase (194 IU/L) and lactate dehydrogenase (1,910 U/L), fasting hypertriglyceridemia (184 mg/dL), hypo-fibrinogenemia (115 mg/dL) with elevated d-dimer (16.6 µg/mL), hyperferritinemia (697 ng/mL), and elevated soluble interleukin-2 receptor level (sIL-2R, 2,626 U/mL). Microbiologic tests came out negative, except for EBV-DNA positivity in the peripheral blood. Bone marrow examination revealed some hemophagocytic histiocytes and a few EBV-encoded-small-nuclear-RNA (EBER) positive cells. Under the impression of MAS, she started methylprednisolone pulse therapy and cyclosporine. The serial EBV levels were decreased in the peripheral blood. Because high fever and thrombocytopenia were not sustainably controlled and liver function was progressively worsening, an IL-1-receptor antagonist anakinra with a high dose of dexamethasone was tried with a prompt fever resolution over several hours. However, the high fever redeveloped during the tapering of dexamethasone, and etoposide was added according to the hemophagocytic lymphohistiocytosis (HLH) 2004 protocol [10]. With the combination therapy with etoposide for 2 months and anakinra for 3 months, her disease was completely controlled (Figure 1). After tapering off the anakinra, she switched to tocilizumab every 4 weeks with cyclosporine as maintenance therapy for AOSD. AOSD and idiopathic urticaria did not recur during the subsequent 12 months.
DISCUSSION
Although AOSD has often been regarded as the adult spectrum of sJIA, there has been no consensus on the cut-off age for classification. JIA traditionally has been defined as arthritis beginning before age 16 years, although recent preliminary criteria defined JIA as disorders appearing before the 18th birthday [11]. Therefore, the current case was classified as AOSD but not sJIA. The diagnosis of MAS is made in the clinical context, based on characteristic features including high persistent fever, cytopenias, NK cell dysfunction with elevated sIL-2R levels, consumptive coagulopathy, and hemophagocytotic macrophage in the tissues [1]. The current case satisfied the HLH-2004 diagnostic criteria and the 2016 classification criteria for MAS in sJIA [10,12].
MAS is one of the life-threatening complications of AOSD with a high mortality rate ranging from 10% to 20% [13] and characterized by the overexpression of proinflammatory cytokines such as IL-1β, IL-6, IL-18, and IFN-γ [1]. The disease flare-ups, infections, and medications are known to be common initiation factors. In the present case, AOSD was treated with tocilizumab and methotrexate and the tocilizumab dose was reduced before MAS development. However, her AOSD had been in remission for 15 months of tocilizumab administered at 6-week intervals. Additionally, CRP production is dependent on IL-6, but her CRP levels were not detected at the current admission. Finally, re-introduction of tocilizumab showed only a transient response. Therefore, tocilizumab dose reduction itself was less likely to provoke MAS. Previously, several cases reported that MAS developed after starting tocilizumab. But, unlike our case, they had active, multi-drug refractory sJIA/AOSD and tocilizumab was administered as an alternative option. Therefore, it is not so much that tocilizumab might trigger MAS but rather that uncontrolled disease could cause MAS in the previous cases [4,6,8].
She started omalizumab for refractory idiopathic urticaria, 5 months before MAS onset. In a literature review, despite no report on omalizumab-induced MAS, omalizumab was reported to increase INF-γ production [14]. IFN-γ can activate macrophages with a proinflammatory phenotype and produce high levels of proinflammatory cytokines [1]. Because these activated macrophages, along with activated CD8+ T cells, have been thought to have a pivotal role in MAS, omalizumab might trigger MAS in the current case. Another possible causative factor included EBV infection whose presence was noted in the peripheral blood and bone marrow. However, she showed no clinical signs of active EBV infection, and during her course, the follow-up EBV levels were decreased.
The most notable feature of this case was that MAS developed during the IL-6 blocking therapy for AOSD. Tocilizumab has been reported to be effective for MAS and refractory AOSD [1,15]. However, MAS that develops upon tocilizumab, could present with some atypical features. In retrospective studies, MAS patients, who developed it upon tocilizumab treatment, had lower WBC counts, lower levels of CRP, ferritin, and fibrinogen, and a higher AST level than those without receiving tocilizumab [3-8]. Our case also showed trends like the features previously reported (Table 1). It should be kept in mind that an IL-6 blocking agent cannot fully prevent the development of MAS and can modify MAS features.
Interestingly, our case shows that MAS could not be fully resolved by a therapy targeting a single cytokine. Anti-IL-1β with anakinra or canakinumab is also accepted as a good therapeutic option for refractory or complicated AOSD and MAS. However, there was an AOSD case complicated by MAS when receiving canakinumab [2]. In our case, MAS developed upon tocilizumab, and it was not sustainably improved by anakinra. According to a recent recommendation based on expert opinion, the initial regimen includes glucocorticoids and anti-cytokine agents (anakinra or tocilizumab) with/without cyclosporine for HLH secondary to autoimmune/inflammatory disease or drugs [15]. In the case of an insufficient response to the initial therapy or central nervous system involvement, etoposide is suggested [15]. Under the tentative diagnosis of MAS, the current case initially received methylprednisolone pulse therapy with cyclosporine. Because the last tocilizumab was received just before the current admission and EBV positivity was reported, there were concerns about the adverse effects of multiple potent immunosuppressants. However, insufficient MAS control finally led to the sequential administration of anakinra and etoposide. Because tocilizumab and anakinra could not reach a sustainable improvement, it supports that many inflammatory cytokines including IL-1β/IL-6 have a role in MAS pathogenesis. Therefore, when MAS is refractory to an anti-cytokine agent, a therapeutic regimen with etoposide should be rapidly commenced for the life-threatening complication of AOSD.
SUMMARY
In summary, we reported the first Korean case with MAS while receiving tocilizumab for AOSD. In diagnosing AOSD/MAS, tocilizumab can modify its typical features. Moreover, because various proinflammatory factors are involved in MAS pathogenesis, therapeutic agents targeting a single cytokine may result in an inadequate response. In patients with multidrug-resistant MAS, the appropriate use of etoposide could successfully manage the disease.
 XML Download
XML Download