

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR: Translocation of platelet-derived growth factor receptor A (PDGRFA)-protein kinase cGMP-dependent 2 (PRKG2) is uncommon in myeloproliferative neoplasms. We report a rare case of a patient presenting Philadelphia-negative myeloproliferative neoplasms with peripheral basophilia. The patient was a 38-year-old man with splenomegaly and leukocytosis. Peripheral blood examination revealed normocytic and normochromic red blood cells, no polychromasia, an increased WBC count with no toxic granulation or vacuolations, some blasts, basophilia, and an increased platelet count. Laboratory tests revealed a white blood cell count of 30,000/mL, hemoglobin level of 11.6 g/dL, and platelet count of 789,000/mL. WBCs comprised 30% segmented neutrophils, 13% lymphocytes, 1% monocytes, 4% eosinophils, and 47% basophils. Bone marrow biopsy showed estimated cellularity of ∼100%, and the numbers of immature cells and basophils were increased. Multifocal fibrosis was also observed. The PDGRFA-PRKG2 fusion gene was observed through next-generation gene sequencing, and targeted imatinib therapy was selected using an artificial intelligence matching algorithm. Immediately after treatment, the patient’s symptoms improved immediately, and his WBC count was normalized. Our results identify the first case of PDGRFA-PRKG2 fusion in an unclassified myeloproliferative neoplasm. This case also shows that the use of imatinib is very effective in treating specific myeloproliferative neoplasms with the PDGRFA-PRKG2 translocation.
INTRODUCTION
Patients, occasionally meet the general criteria for classifying their condition as a myeloproliferative neoplasm (MPN) but may not meet all criteria for a particular disease or may exhibit more than one category of diagnostic characteristics. These patients can be diagnosed as MPN unclassifiable (MPN-u). Symptoms are similar to typical MPNs and usually include hepatosplenomegaly and increased numbers of white blood cells (WBCs) and platelets. A bone marrow biopsy reveals megakaryocyte proliferation and hypercellularity in granulocytes or erythrocytes. As the disease progresses, the bone marrow becomes more fibrotic. However, in the current patient, BCR-ABL, CARL, MPL, and JAK2 gene mutations, commonly observed in MPNs, were all negative. The patient was treated with hydroxyurea and ruxolitinib due to leukocytosis, hyperbasophilia, and splenomegaly, but there was no improvement. Myeloproliferative diseases often have mutations or rearrangements of protein tyrosine kinase genes such as JAK2, FGFR1, platelet-derived growth factor receptor A (PDGFRA), and PDGFRB [1, 2]. A variety of chromosomal translocations involving PDGFRA have been identified revealing that it can fuse with many different partner genes. PDGFRA and PDGFRB are transmembrane glycoproteins in the type III receptor tyrosine kinase family. This family also contains KIT, FLT3, and c-FMS. Platelet-derived growth factors (PDGFs) include a group of mitogens comprising five dimeric forms derived from pairs of A, B, C, and D peptide chains (PDGF-AA, PDGF-AB, PDGF-BB, PDGF-CC, and PDGF-DD). PDGFRA binds to the A, B, and C chains. PDGFRB binds to B and D chains. PDGFRA and PDGFRB are transmembrane glycoproteins belonging to the type III receptor tyrosine kinase family [3]. In 2003, the FIP1L1-PDGFRA mutation was described in relation to the MPN phenotype characterized by eosinophilia and mastocytosis [4]. The FIP1L1-PDGFRA fusion gene was highly sensitive to low-dose imatinib; complete molecular remission could be achieved with 100 mg/day and a 300 mg/week maintenance dose [5]. To date, four PDGFRA fusion genes have been identified including FIP1L1-PDGFRA, BCR-PDGFRA, ETV6-PDGFRA, and CDK5RAP2-DGFRA by the nested real-time polymerase chain reaction [6-8]. However, these mutations have been reported to exhibit clinical patterns of hyperbasophilia. In this study, we report a case of MPN-u associated with basophilia and the protein kinase cGMP-dependent 2 (PRKG2)-PDGFRA translocation, which was improved following imatinib treatment.
CASE
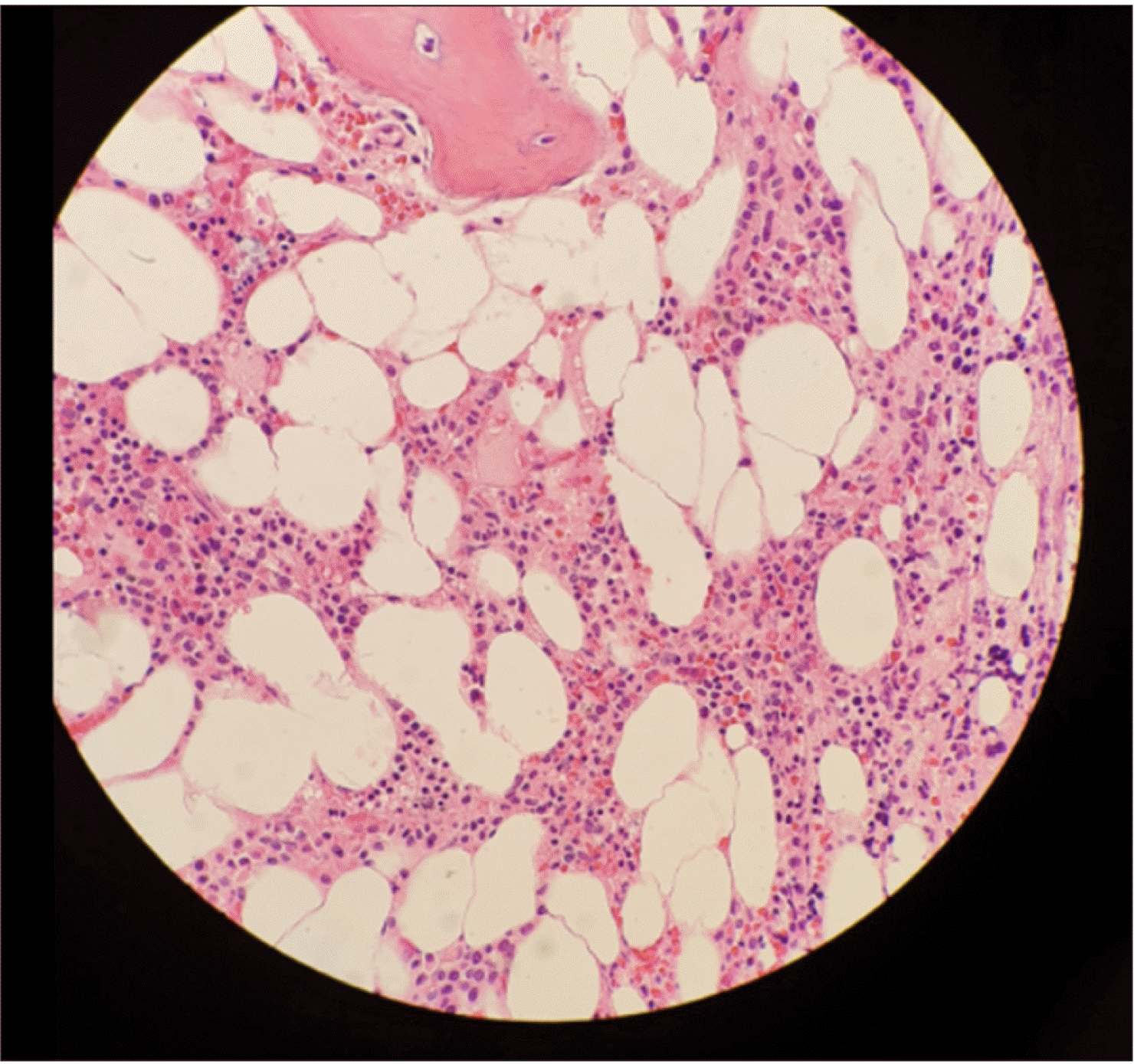
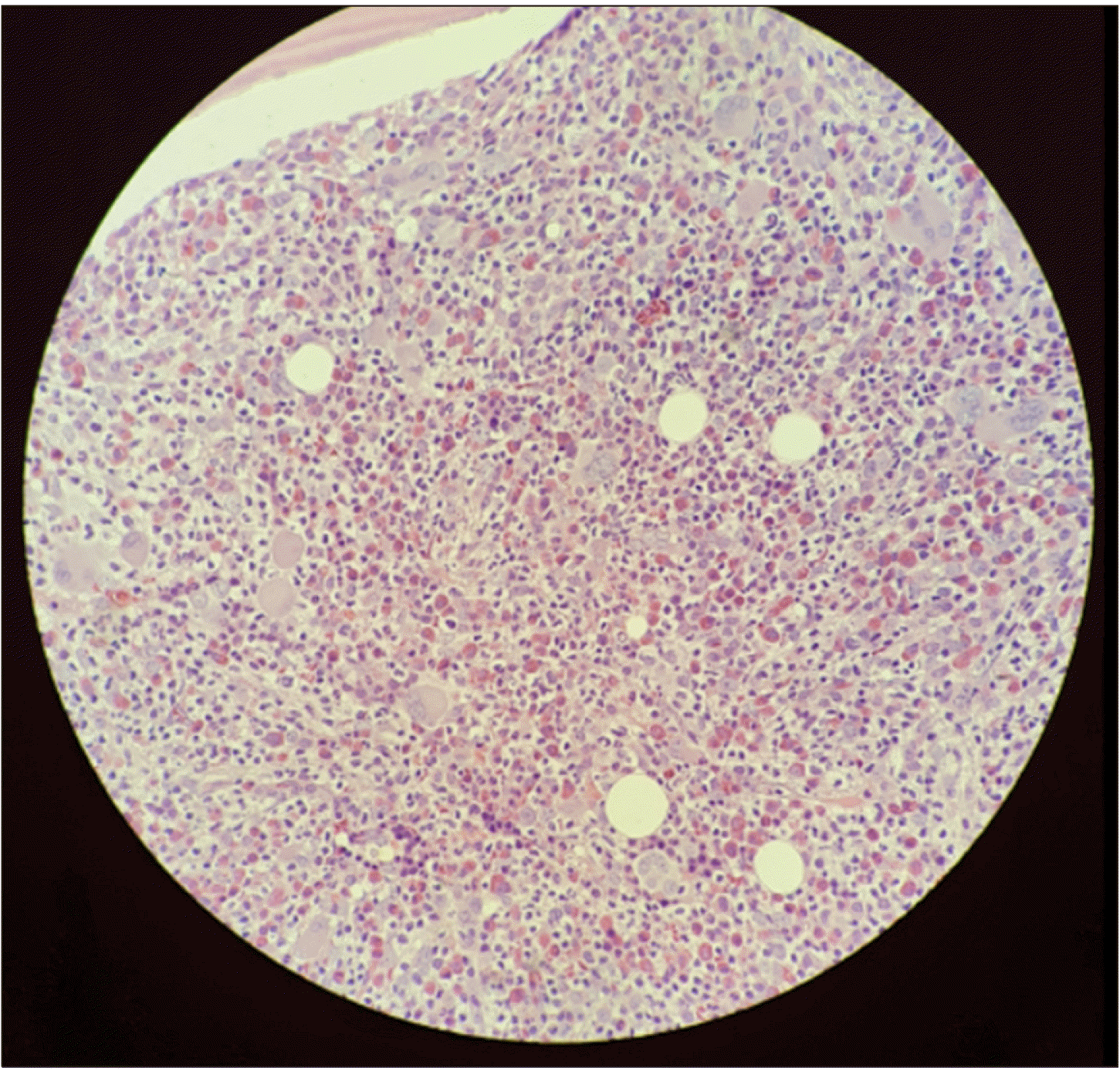
A 38-year-old man presented to the Konyang University Hospital (Deajeon, South Korea) on March 10, 2017, with abdominal pain that started 2 months before, and thrombocytosis was identified at a local clinic. At the time of admission, the patient had a blood pressure of 130/90 mmHg, heart rate of 78 beats per minute, respiratory rate of 20 breaths per minute, and body temperature of 37°C. There was no significant previous medical history of hypertension, diabetes mellitus, or tuberculosis. On physical examination, we observed organomegaly in the right upper quadrant of the abdomen. However, there was no enlargement of the cervical, axillary, or inguinal lymph nodes. Other examinations were unremarkable. Laboratory tests showed WBCs at 30,300/mL, hemoglobin at 9.4 g/dL, and platelets at 836,000/mL. White blood cells were composed of 24% segmented neutrophils, 17% lymphocytes, 1% monocytes, 3% eosinophils, and 51% basophils. The reticulocyte count was within the normal range. Leukemic blasts were observed in 2% of the cells. Coagulation tests revealed a prothrombin time of 16.4 seconds and an activated partial thromboplastin time of 35.5 seconds. He had a fibrinogen level of 3.13 g/L, D-dimer level of 2.1 mg/mL, and antithrombin activity of 72% (not suggestive of disseminated intravascular coagulation). Peripheral blood showed normocytic and normochromic red blood cells, no polychromasia, a normal WBC count with no toxic granulation or vacuolations, and an increased platelet count. The serum lactate dehydrogenase level was 832 U/L (normal range, 120–240 U/I). The total bilirubin level was 1.04 mg/dL (normal range, 0–0.4 mg/dL). Results for other blood factors, including creatinine and bicarbonate, and liver function tests, were unremarkable. Computed tomography revealed hepatosplenomegaly (16 cm) without an intrasplenic mass. Bone marrow aspiration revealed no particles or peripheral dilution, and immature cells were observed occasionally (Fig. 1). Bone marrow biopsy showed estimated cellularity of approximately 100%, which was hypercellular for the patient’s age. In cellular areas, trilineage hematopoiesis was observed, along with increased numbers of basophils and immature cells. In addition, there were increased numbers of dysplastic megakaryocytes, and diffuse fibrosis was observed in multiple focal areas throughout the bone marrow space (Fig. 2). The JAK2 V617F, and BCR/ABL1 mutations were not detected with the real-time polymerase chain reaction. In chromosome analysis, of the 20 metaphase cells that could be observed, all cells were subjected to 46, XY normal karyotype. The initial formal pathologic diagnosis was deferred, so the patients received hydroxyurea 200 mg twice daily. However, leukocytosis continued, and the basophilia did not improve. The WBC count eventually increased to 88,000/mL, and a bone marrow biopsy was performed again. The second formal pathologic diagnosis was MPN with myelofibrosis. The patient additionally received 20 mg ruxolitinib phosphate twice daily. Although the WBC count decreased to 32,000/mL, basophilia persisted. Various symptoms, such as splenomegaly, basophilia, and leukocytosis continued after more than a year of treatment. Finally, the patient was able to undergo next-generation gene sequencing (NGS). Targeted sequencing was performed for translocation detection using the HemaSCAN panel (Level.1), which includes the whole exomes of 427 cancer-associated genes, and the intronic regions of 23 genes. The sequencing library was constructed with DNA extracted from bone marrow aspiration blood, and sequencing was performed on a NextSeq550Dx with a 75 bp read (Illumina). Post-sequencing data were analyzed using a bioinformatics pipeline (HemaSCAN version 1.0) from Labgenomics. This test identified a driver mutation, that is, the PRKG2-PDGFRA translocation (Table 1). Based on artificial intelligence matching algorithm (HemaSCAN version 1.0), imatinib treatment was immediately initiated. Within a month of treatment, the WBC level was within the normal range, and the basophil level was reduced to 1%. The patient was treated with imatinib for more than a year, and the PDGRFA-PRKG2 translocation disappeared in a repeated NGS test (Table 2). All of the patient’s blood tests results returned to normal and splenomegaly improved.
DISCUSSION
Translocation of PDGRFA-PRKG2 is uncommon in MPNs. Here, we report a rare case of a patient presenting with a Philadelphia-negative MPN with peripheral basophilia. These results are the first report of PDGRFA-PRKG2 fusion in an MPN-u case. This case shows that the use of imatinib is very effective in specific MPNs with the PDGRFA-PRKG2 translocation. The most intensively studied PDGFRA mutation is FIP1L1-PDGFRA, a karyotypically occult del(4)(q12) described in 2003 as an imatinib-sensitive activation mutation. The FIP1L1-PDGFR fusion gene is an ∼800 kb interstitial deletion within 4q12 that fuses the 5′ portion of FIP1L1 to the 3′ portion of PDGFRA and is a novel molecular mechanism for generating a compositionally activated fusion tyrosine kinase. The breakpoint of FIP1L1 has been observed at various positions, but the PDGFRA breakpoint is specific to exon 12, which encodes part of the protein-protein interaction module with two fully conserved tryptophans containing the JM region. As a result, automatic prohibition activities are suspended, leading to disruption of its autoinhibitory activity [9]. Although FIP1L1-PDGFRA occurs in a small number of patients with phenotypic features of systemic mastocytosis or hypereosinophilic syndrome, the presence of this mutation reliably predicts complete hematological and molecular responses to imatinib therapy [10]. In addition, PDGFRA activation associated with chronic eosinophilic leukemia is described by karyotypically apparent fusion mutations such as KIF5B-PDGFRA t(4:10)(q12;p11) [11], BCR-PDGFRA, t(4;22)(q12;q11) [12], and CDK5RAP2-PDGFRA, ins(9;4) (q33;q12q25) [13].
KIF5B is the third PDGFRA partner gene identified after BCR and FIP1L1, and encodes kinesin family member 5b, a microtubule-based motor protein involved in organ transport. The KIF5B protein is composed of three structural regions: an N-terminal region that hydrolyzes ATP and binds microtubules, a large central helicoil region, and a C-terminal region that interacts with other proteins, blood vessels, and membrane organs. The KIF5B-PDGFR fusion protein contains most of the first two structural domains of KIF5B, including six of the seven coil domains and the entire kinase domain of PDGFRA [11].
CDK5 regulatory subunit-related protein 2 (CDK5RAP2) encodes a protein that is thought to be involved in regulating the formation and stability of microtubules from the centrosome. A new mRNA in-frame fusion between exon 13 of the CDK5RAP2 gene, and truncated PDGFRA exon 12, were identified by rapid amplification of cDNA ends with the polymerase chain reaction. The CDK5RAP2-PDGFRA protein, which is composed of 1,003 amino acids, preserves both the tyrosine kinase domain of PDGFRA and several potential dimerization domains of CDK5RAP2 [13]. In 1994, the Golub and Gililand groups described ETV6-PDGFRB fusion as the first of these fusion genes in patients with chronic myelomonocytic leukemia with eosinophilia and t(5;12) [14]. This gene encodes a protein that belongs to the serine/threonine protein kinase family of proteins. The encoded protein then binds to and inhibits the activation of several receptor tyrosine kinases. Alternate splicing results in multiple transcript variants encoding distinct isoforms whose regulatory N-termini differ in length but whose C-terminal catalytic domains are identical.
The PRKG2 gene encodes a protein that belongs to the serine/threonine protein kinase family of proteins. The encoded protein binds to and inhibits the activation of several receptor tyrosine kinases and plays a role in regulating fluid balance in the intestine. Membrane-bound proteins are regulators of the intestinal secretion, bone growth, and renin secretion. Diseases associated with PRKG2 include chromosome 4Q21 deletion syndrome and cystic fibrosis. Among its related pathways are Sertoli-Sertoli cell junction dynamics and signaling by G-protein coupled receptors [15]. The PRKG2-PDGFRB fusion gene associated with t(4;5)(q21;q33) has been reported previously in only a few patients that presented with systemic mastocytosis, basophilia, thrombocytosis, and massive splenomegaly, with a myeloid neoplasm and PRKG2-PDGFRB rearrangement [16]. Our case, together with those cases in the literature, suggests an association between PDGRFA-PRKG2 and basophilia without mastocytosis. Our patient responded well to imatinib therapy. Further studies are needed to determine if the PDGRFA-PRKG2 fusion is harbored by basophils, which would suggest that cases may represent a distinct clonal disorder. In conclusion, this case highlights the fact that patients with PDGRFA-PRKG2 fusion respond favorably to imatinib. Genetic analysis by NGS in ambiguous blood cancers is essential for identifying appropriate targeted treatment, and further research is warranted.
 XML Download
XML Download