

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Philadelphia chromosome-negative myeloproliferative neoplasms (Ph- MPNs) include essential thrombocythemia (ET), polycythemia vera (PV), prefibrotic primary myelofibrosis (pre-PMF), and overt primary myelofibrosis (PMF) [1]. In addition to thrombotic and hemorrhagic vascular events, life-threatening myelofibrotic and leukemic transformations are the main clinical manifestations of Ph- MPNs. The incidence and clinical features of disease transformation are based on reports from Western countries, and data from Asian populations are scarce [2, 3]. Although a few Korean studies on MPNs [4] have been conducted, the available data are relatively limited. The diagnostic criteria for Ph- MPNs have frequently been revised [1, 5-7]. The diagnostic thresholds of hemoglobin and hematocrit for PV and platelet counts for ET have been reduced considerably, and pre-PMF is defined more clearly by the diagnostic criteria proposed by the World Health Organization (WHO) [1]. Therefore, some patients previously diagnosed with ET are now classified as patients with pre-PMF or PV [8-12], which necessitates the reanalysis of data on MPN transformation. The increasing incidence of MPN in Korea [13-15] is partly attributable to changes in diagnostic criteria and studies on driver gene mutations, such that there is a need for new data on disease transformation in these patients. In this retrospective study, we analyzed myelofibrotic and leukemic transformations in Korean patients diagnosed with Ph- MPN, based on the 2016 WHO criteria [1].
MATERIALS AND METHODS
Patient recruitment and acquisition of data
Patients diagnosed with ET, PV, pre-PMF, or PMF between January 1996 and December 2020 at the Chungnam National University Hospital, Daejeon, Korea, were enrolled in this study. Demographic and laboratory data were extracted from medical records, including complete blood counts and blood chemistry values, driver gene mutations, bone marrow (BM) examination results, and cytogenetic study results. The International Prognostic Score for Essential Thrombocythemia (IPSET) [16] and International Prognostic Scoring System (IPSS) [17] were used for the prognostic stratification of patients with ET and PMF, respectively. For patients diagnosed with ET before 2017, the diagnoses were revised based on the 2016 WHO diagnostic criteria [1]. All laboratory data and BM specimens generated at the initial diagnosis were reviewed and re-examined through the collaboration of a hematologist and hematologic pathologist. Hydroxyurea or anagrelide was used for cytoreduction based on the standard recommendations, drug availability, and compliance. Low-dose aspirin (100 mg/day) was prescribed to prevent thrombosis, except for low- and very-low-risk patients.
Driver gene mutation analyses
The Janus kinase 2 mutation (JAK2V617F) was identified using polymerase chain reaction (PCR) and Sanger sequencing (before 2010) and allele-specific real-time quantitative PCR (after 2010). A calreticulin (CALR) mutation in exon 9 was detected using fragment analysis and Sanger sequencing. Myeloproliferative leukemia gene mutation (MPLW515K/L) was assessed using PCR and Sanger sequencing.
Definitions of myelofibrotic and leukemic transformations
Myelofibrotic transformation refers to secondary myelofibrosis (SMF) that develops in ET (post-ET myelofibrosis, PET-MF) or PV (post-PV myelofibrosis, PPV-MF) patients, and the progression of pre-PMF to PMF. Myelofibrotic transformation was diagnosed based on the 2016 WHO criteria [1]. Leukemic transformation (to acute myeloid leukemia) was defined as the presence of ≥20% blasts in peripheral blood or BM.
Definitions of thrombotic events
Thrombotic events included cerebrovascular (ischemic stroke, transient ischemic attack, and venous sinus thrombosis), coronary (any ischemic heart disease, including acute coronary syndrome), splanchnic, and peripheral thrombo-embolism.
Statistical analysis
Descriptive data are presented as mean±standard deviation (SD), median (range), or percentage and were compared using Student’s t-test, chi-squared test, or Fisher’s exact test. The cumulative incidence of myelofibrotic and leukemic transformation according to the MPN subtype was calculated using the Fine and Gray model, with death serving as a competing risk, and analyzed using the Gray equality test. Risk factors for transformation were analyzed using the Fine and Gray regression model, with death serving as a competing risk. Overall survival (OS) was defined as the time from MPN diagnosis to death from any cause. Survival was estimated using the Kaplan-Meier method and analyzed using the log-rank test. Statistical analyses were performed using the SPSS software (version 24.0, IBM Corp., Armonk, NY, USA) or SAS University Edition (SAS Institute, Cary, NC, USA). Statistical significance was set at P-value <0.05.
RESULTS
Patient characteristics
A total of 351 patients (144 with ET, 131 with PV, 45 with pre-PMF, and 31 with PMF; 204 men and 147 women) with a median age of 64 years (range, 15–91 years) were enrolled. They were followed for a median of 4.6 years (range, 0.2–24.8 years). Patients with ET diagnosed before 2017 were rediagnosed based on the 2016 WHO diagnostic criteria. Of the 129 patients with ET, 32 (24.8%) and 11 (8.5%) were reclassified as pre-PMF and PV, respectively. Palpable splenomegaly was most frequently observed in patients with PMF (51.6%), followed by patients with pre-PMF (8.9%) and PV (8.4%). None of the patients with ET exhibited palpable splenomegaly. White blood cell (WBC), monocyte, and platelet counts and lactate dehydrogenase (LDH) normalized ratio were higher in patients with pre-PMF than in patients with ET [14.5±10.2×109/L and 11.0±4.5×109/L; 0.8±0.4×109/L and 0.6±0.4×109/L; 1,093.9±461.1×109/L and 946.5±244.9.4×109/L; and 1.6±0.7×upper normal limit (UNL) and 1.1±0.4×UNL, respectively; all P<0.05]. JAK2V617F was most common in patients with PV (87.7%), followed by patients with ET (68.0%), PMF (65.3%), and pre-PMF (63.2%). CALR mutations were most frequently found in patients with PMF (21.7%), followed by patients with pre-PMF (13.2%) and ET (11.5%). Patients with ET were evenly distributed among the IPSET risk groups. Most patients with pre-PMF were in the low (53.3%) or intermediate-1 (37.8%) IPSS risk group, while most patients with PMF were in the intermediate-2 (38.7%) or high (19.4%) risk groups. Cytoreductive therapy was most commonly prescribed for patients with PV (82.4%), followed by patients with ET (75.7%), pre-PMF (75.6%), and PMF (29.0%). Except for patients with PMF, the majority of patients with MPN were placed on low-dose aspirin (Table 1).
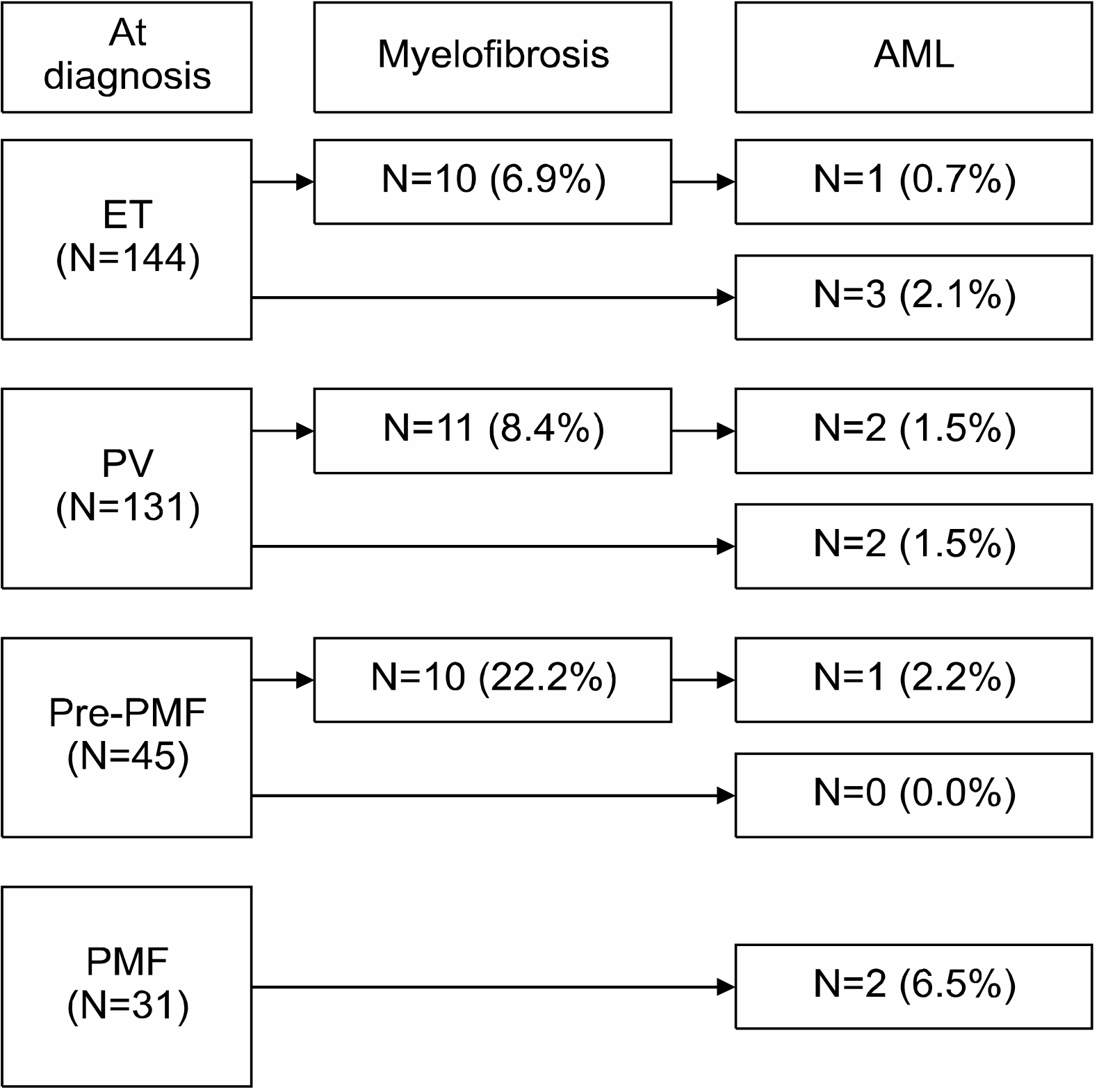
Prevalence and cumulative incidence of myelofibrosis and leukemia
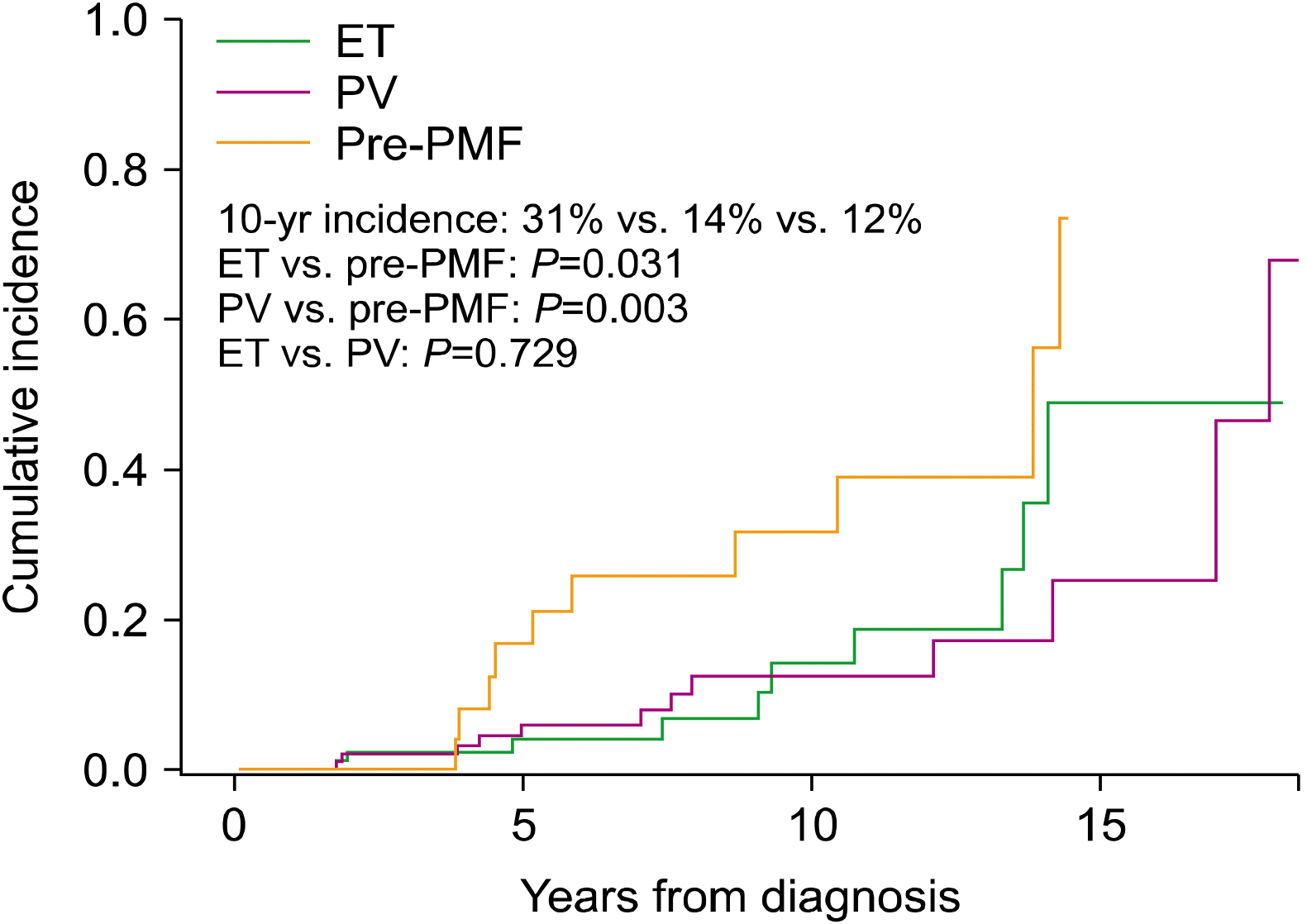
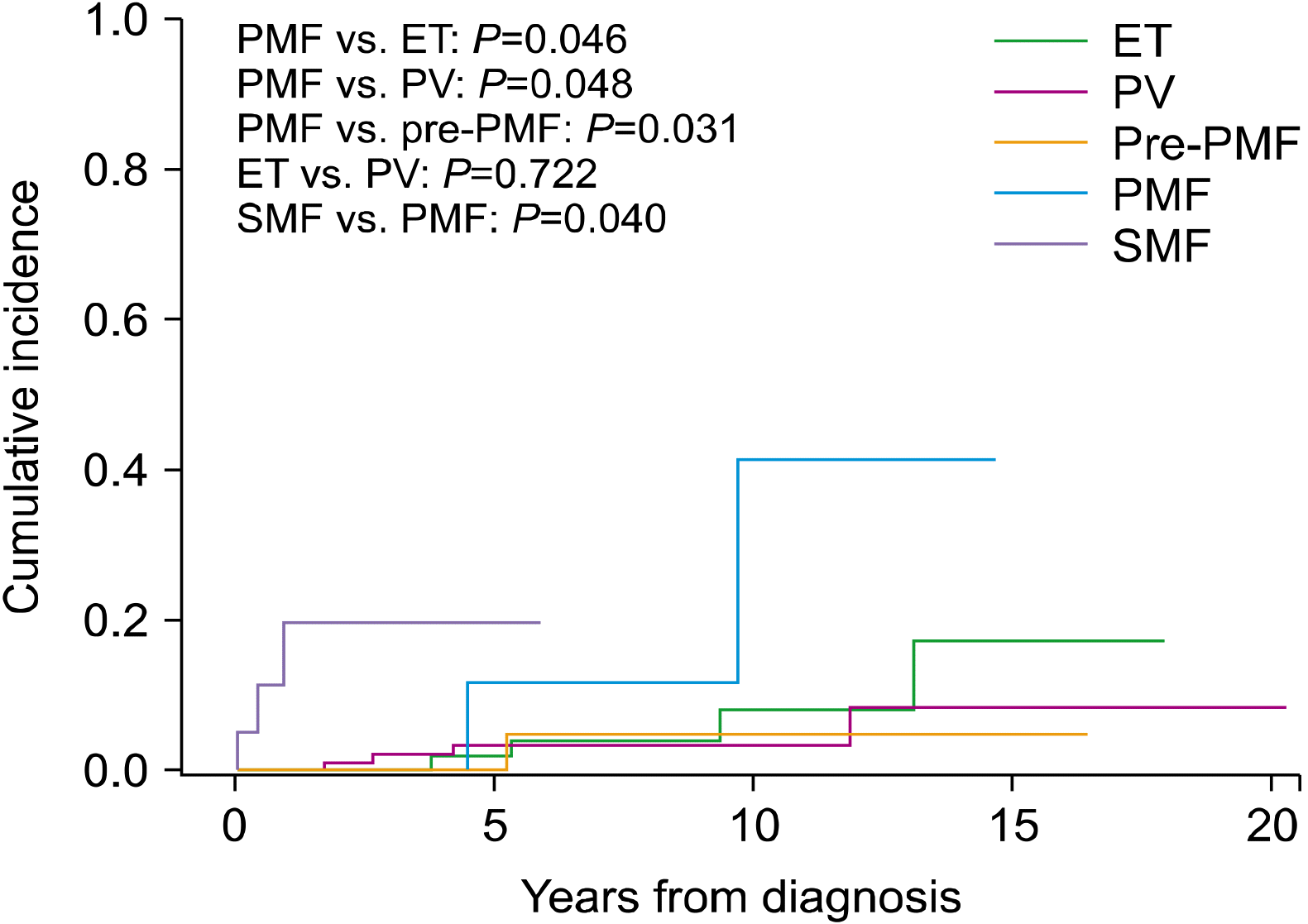
To determine myelofibrotic transformation, 33 BM examinations were performed in 31 patients (10 of 10 patients with ET, 13 of 11 patients with PV, and 10 of 10 patients with pre-PMF). Myelofibrotic transformation was most common in patients with pre-PMF (22.2%), followed by patients with PV (8.4%) and ET (6.9%). Leukemic transformation was most common in patients with PMF (6.5%), followed by patients with PV (3.0%), ET (2.8%), and pre-PMF (2.2%) (Fig. 1). The cumulative incidence of myelofibrotic transformation was significantly higher in patients with pre-PMF than in patients with ET (8-year incidence, 25.5% and 6.5%; 10-year incidence, 31.3% and 13.7%, respectively; P=0.031) and PV (8-year incidence, 12.2%; 10-year incidence, 12.2%; P=0.003), while no differences were observed between patients with ET and PV (P=0.729). The 15-year cumulative incidence rates of myelofibrotic transformation were 48.4% and 24.9% in patients with ET and PV, respectively (Fig. 2). The cumulative incidence of leukemic transformation was significantly higher in patients with PMF than in patients with ET (8-year incidence, 11.4% and 3.8%; 10-year incidence, 40.0% and 7.9%, respectively; P=0.046), pre-PMF (8-year incidence, 4.7%; 10-year incidence, 4.7%; P=0.048), and PV (8-year incidence, 3.2%; 10-year incidence, 3.2%; P=0.031). The 15-year cumulative incidence rates of leukemic transformation were 16.9%, 8.2%, and 4.3% in patients with ET, PV, and pre-PMF, respectively. The cumulative incidence of leukemic transformation in patients with SMF was significantly higher than in patients with PMF (5-year incidence, 19.0% and 11.4%, respectively; P=0.040) (Fig. 3).
Clinical features of patients with SMF
The clinical features of patients at the time of PET-MF and PPV-MF diagnosis were compared to those at the initial ET and PV diagnosis, respectively. Among 10 patients with PET-MF, 6 (60.0%) developed palpable splenomegaly. No differences were noted in the WBC or monocyte counts between the two groups, but the hemoglobin levels (12.9±2.5 g/dL and 8.9±1.5 g/dL, respectively; P=0.001) and platelet counts (667.0±84.57×109/L and 527±486.8×109/L, respectively; P=0.003) were significantly lower, and the LDH normalized ratio (1.3±0.6×UNL and 3.8±1.2×UNL, respectively; P=0.004) were significantly higher, at the time of PET-MF diagnosis. Leukoerythroblastosis and abnormal karyotypes were newly found in 8 (80.0%) and 3 (30.0%) patients with PET-MF, respectively. Of the 11 patients with PPV-MF, 1 (9.1%) had newly developed palpable splenomegaly. Although no differences were noted in the WBC, monocyte, or platelet counts between the two groups, hemoglobin levels (17.3±3.1 g/dL and 10.6±1.4 g/dL, respectively; P=0.001) were significantly lower, and LDH normalized ratio (1.1±1.1×UNL and 2.2±0.6×UNL, respectively; P=0.037) were significantly higher at the time of PPV-MF diagnosis. Leukoerythroblastosis and abnormal karyotypes were newly found in 10 (90.9%) and 4 (36.4%) patients with PPV-MF, respectively (Table 2).
Clinical features of PMF progressed from pre-PMF
The clinical features of patients with PMF who progressed from pre-PMF at diagnosis were compared to those at the initial pre-PMF diagnosis. Among 10 patients with PMF, 5 (50.0%) had newly developed palpable splenomegaly. Whereas no differences were noted in WBC and monocyte counts between the two groups, hemoglobin levels (13.2±2.4 g/dL vs. 9.2±2.5 g/dL, P=0.004) and platelet counts (1,186.3± 567.7×109/L vs. 450.7±196.5×109/L, P=0.003) were significantly lower, and LDH normalized ratio (1.5±0.4×UNL vs. 2.8± 0.5×UNL, P=0.005) was significantly higher at the time of overt PMF diagnosis. Among 10 patients with PMF, 7 (70.0%) had newly developed leukoerythroblastosis, and 6 (60.0%) newly exhibited abnormal karyotypes. In terms of IPSS risk stratification, more patients belonged to intermediate-2 or high-risk groups after myelofibrotic transformation (0.0% vs. 80.0%, P=0.004) (Table 3).
Clinical features of SMF and leukemia
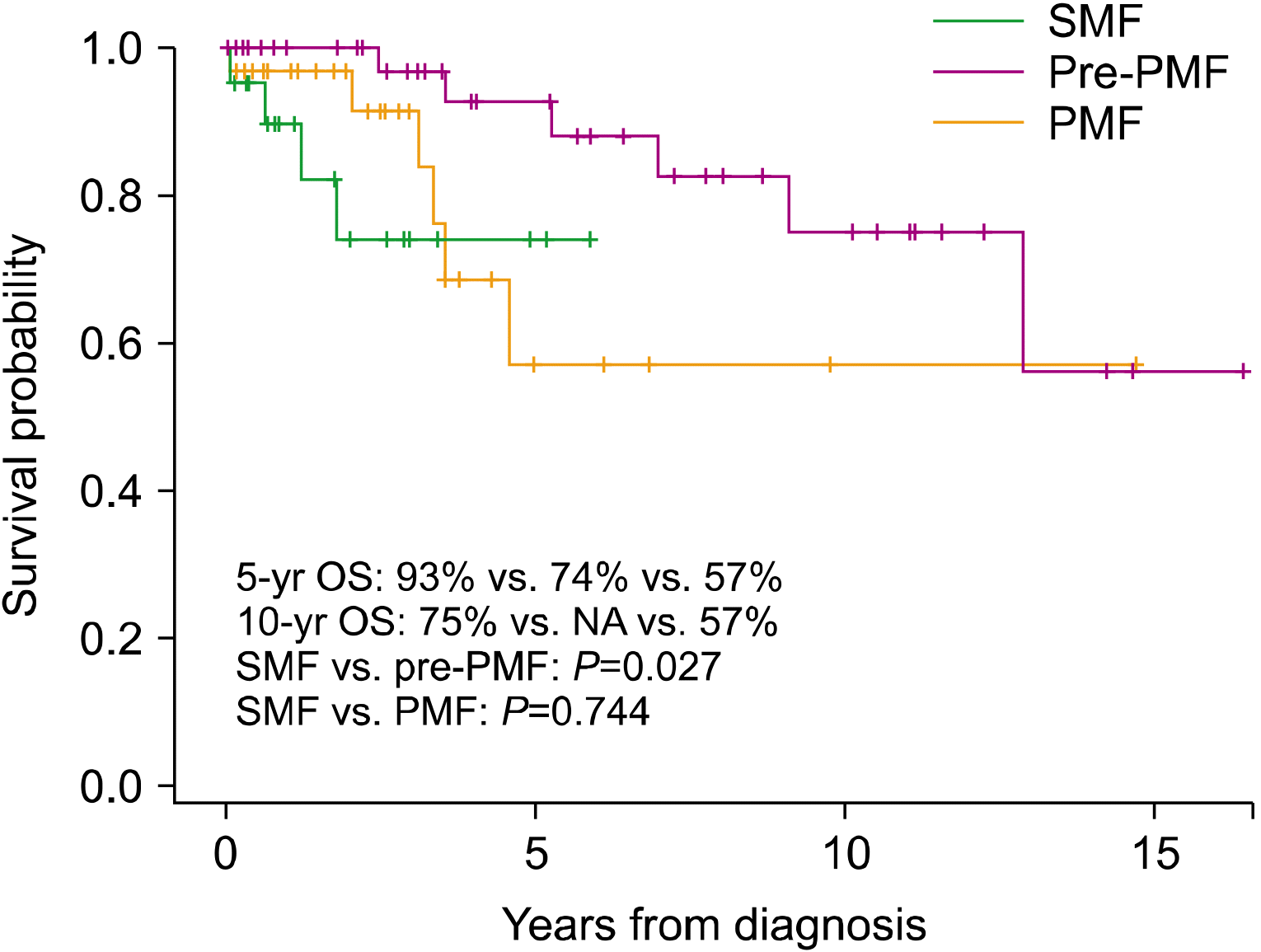
The clinical features of SMFs (N=21) at diagnosis were compared with those of PMFs (N=31). The groups had similar rates of palpable splenomegaly and similar WBC counts, hemoglobin levels, platelet counts, and LDH normalized ratios. Abnormal karyotypes were more common in patients with SMF than in patients with overt PMF (45.0% and 9.7%, respectively; P=0.002). More patients with SMF were classified into the intermediate-2 or high-risk IPSS group than into PMF group (90.0% and 58.1%, respectively; P=0.038) (Table 4). The 5-year OS was significantly lower in patients with SMF than in those with pre-PMF (74% and 93%, respectively; P=0.027) but did not differ from that in patients with PMF (57%; P=0.744) (Fig. 4). The prognosis of patients with secondary leukemia (N=11) was dismal, with a median survival of 1.5 months (range, 0.5–14.5 mo) (data not shown).
Risk factors for SMF and leukemia
Fine and Gray regression analyses were performed to determine the risk factors for SMF in patients with ET and PV. In patients with ET, high monocyte counts (>1.0×109/L) [hazard ratio (HR), 3.57; 95% confidence interval (CI), 1.17–10.91; P=0.026] and CALR mutations (HR, 4.42; 95% CI, 1.20–16.37; P=0.026) at the time of diagnosis were independent risk factors (Table 5). In patients with PV, abnormal karyotypes (HR, 18.20; 95% CI, 2.0–165.95; P=0.010) at the time of diagnosis was a sole independent risk factor (Table 6). Hydroxyurea treatment and thrombosis did not affect myelofibrotic transformation in patients with ET and PV. Fine and Gray regression analyses were also performed to determine the risk factors for leukemia in patients with MPN. High monocyte counts (>1.0×109/L) (HR, 4.05; 95% CI, 1.23–13.39; P=0.022) and abnormal karyotypes (HR, 5.60; 95% CI, 1.10–28.59; P=0.038) were risk factors in univariate analysis; however, the statistical significance was lost in multivariate analysis. Driver gene mutations, MPN type, and hydroxyurea treatment did not affect leukemic transformation (Table 7).
DISCUSSION
Western studies have reported 1.6–9% and 5–14% 10–15-year cumulative incidence rates of myelofibrotic transformation in patients with ET and PV, respectively [18-22]. In the present study, the 10-year cumulative incidence rates of SMF in patients with ET and PV were 13.7% and 12.2%, respectively, similar to those in Western studies. A recent Korean study based on nationwide public healthcare insurance claims data reported 8-year cumulative SMF incidence rates of 2.8% and 1.2% among 4,307 and 2,470 patients with ET and PV, respectively [4], which were significantly lower than in the present study. However, that large data study analyzed only public health insurance claims data; individual medical records were not reviewed. Therefore, the incidence rates may have been underestimated even though pre-PMF was not differentiated from ET. SMF was diagnosed by BM examination. The timing of BM studies is physician-dependent, and there are significant variations between physicians and institutions. In addition, it is difficult to persuade patients to undergo BM examination. These factors may have influenced the results of this study. An early Chinese study of 231 patients with ET reported a 9.7% probability of myelofibrotic transformation over 10 years [2]. Altogether, the probability of myelofibrotic transformation in patients with ET and PV seems to be similar between the Asian and Western populations. In our study, palpable splenomegaly was commonly observed during SMF diagnosis in patients with ET. Anemia, leukoerythroblastosis, and elevated LDH levels are common features of SMF in patients with ET and PV. These clinical features are known indicators of myelofibrotic transformation and are useful determinants of the timing of BM examinations [23, 24].
The risk of leukemic transformation varies greatly with the features at the time of diagnosis, with the 10-year risk being highest in PMF (10-year risk, 10–20%), followed by PV (2–4%) and ET (1–2%) [21, 22, 25-27]. A few studies have addressed this in Asian populations, but the data are limited, and comparisons with Western studies are difficult [2, 28, 29]. A Korean study reported 8-year cumulative incidence rates for leukemic transformation of 3.6%, 1.7%, and 21.4% in patients with ET, PV, and PMF, respectively [4]. In the present study, the 8-year cumulative incidence rates of leukemic transformation were 3.8%, 3.2%, and 11.4% in ET, PV, and PMF, respectively. Although the cumulative incidence rates slightly increased over time in patients with ET and PV, a marked increase was observed in patients with PMF (up to 40.0% over 10 years). The incidence rate may have been overestimated because of the small number of patients and relatively few transformation events a long time after PMF diagnosis. Taken together, these data suggest that the probability of leukemic transformation in Korean populations does not differ from that in Western populations.
A large international study highlighted the prognostic relevance of distinguishing between pre-PMF and ET in terms of overt PMF transformation risk [21]. Therefore, it is intuitive that the cumulative incidence of overt PMF in patients with pre-PMF was significantly higher than in patients with ET in the present study. These findings emphasize the need for caution when interpreting data from early studies that did not distinguish between pre-PMF and ET. In this study, the clinical features of patients at the time of pre-PMF diagnosis, including palpable splenomegaly, laboratory findings, and IPSS score, differed significantly from those at the time of PMF diagnosis. Most notably, the frequency of cytogenetic abnormalities increased from 10% to 70%, indicating disease progression associated with clonal evolution.
Evidence suggests that patients with PET-MF and PPV-MF differ from patients with PMF in terms of clinical features and outcomes [30, 31]. In accordance with the need to improve prognostic scoring systems for PET-MF and PPV ET, myelofibrosis secondary to PV and the ET-Prognostic Model (MYSEC-PM) was introduced [32]. In the present study, no differences were found between SMFs and PMFs in palpable splenomegaly, CBC profiles, LDH levels, or driver gene mutations. However, more patients with SMF had cytogenetic abnormalities, and more belonged to higher risk IPSS groups than patients with PMF. The cumulative incidence of leukemic transformation in patients with SMF was higher than that in patients with PMF, which contradicts the MYSEC data [32]. No statistically significant differences were found between patients with SMF and PMF in the survival analysis. However, patients with SMF tend to die earlier than patients with PMF, most likely due to leukemic transformation. These observations suggest that SMF diagnosis was delayed; that is, it was not made until the disease had fully progressed, where early diagnosis might have changed the outcomes.
In this study, monocytosis and CALR mutations at the time of ET diagnosis were independent risk factors for myelofibrotic transformation. CALR mutation is associated with a higher risk of myelofibrotic transformation than JAK2 and MPL mutations [33, 34]. However, the risk associated with monocytosis was unexpected. Leukocytosis, palpable splenomegaly, and BM reticulin fibrosis at the time of diagnosis were associated with myelofibrotic transformation in patients with PV [23]. The present study found that an abnormal karyotype at diagnosis was the sole independent risk factor for MF evolution. These results need to be validated in studies that include a larger number of Korean patients. Interestingly, monocytosis was a risk factor for leukemic transformation in our univariate analysis of MPNs, although the statistical significance was lost in the multivariate analysis. The role and implications of monocytosis at diagnosis in terms of myelofibrotic and leukemic transformation need to be further explored. Leukemogenic effects of HU were not observed in the present study. The leukemogenic potential of HU remains controversial because of the difficulties in performing large prospective randomized trials [35]. This study was retrospective and included a limited number of patients. In addition, a subpopulation of patients were followed for a relatively short period. Therefore, the results of this study should be verified in a prospective study involving a large number of patients.
In summary, the prevalence and cumulative incidence of myelofibrotic and leukemic transformation in Korean patients with Ph MPN did not differ from those in Western populations. The prognostic implications of monocytosis at MPN diagnosis in terms of myelofibrotic and leukemic transformation require further investigation.
 XML Download
XML Download