

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The mutational and epigenetic landscapes of acute myeloid leukemia (AML) have become increasingly well understood in recent years, informing on biological targets for precision medicine [1-3]. Among the most notable findings was the recognition of mutational hot-spots in the isocitrate dehydrogenase (IDH) genes. The IDH family comprises of three isozymes (IDH1, IDH2 and IDH3). IDH1 and IDH2 are homo-dimeric enzymes encoded for by the IDH1 and IDH2 genes located on chromosomes 2q33 and 15q26, respectively. IDH2 is localized in the mitochondria, while IDH1 is in the cytosol and peroxisome [4]. The third isozyme, IDH3, is localized in the mitochondria and composed of three distinct subunits encoded for by three different genes (IDH3A, IDH3B, IDH3G). Normally, IDH1/2 enzymes catalyze the oxidative decarboxylation of isocitrate to a-ketoglutarate (a-KG) to produce reduced nicotinamide adenine dinucleotide phosphate (NADPH) from NADP+, and also constitute an important component of the tricarboxylic acid cycle [5]. As many dioxygenases depend on adequate levels of a-KG for various cellular processes [6], alterations in IDH1/2 functions interfere with detoxification mechanisms, thus causing DNA damage and genome instability [7].
In this review article, we discuss IDH1/2 mutations in Korean AML patients, and their potential application as biomarkers and drug targets.
IDH mutations in AML
Overall, IDH-mutant leukemia represents approximately 20% of all AML cases. Clinically, patients with IDH mutations tend to be older with median age of onset at 67 years, and have higher platelet count, bone marrow and peripheral blast counts and more profound neutropenia at the time of diagnosis [8]. In addition, IDH mutations are mostly found in intermediate-risk groups, especially in the normal karyotype (NK) [9].
Biologically, IDH1/2 mutations are predominantly somatic and rarely germline [10], and nearly always cause a single amino acid substitution in Arg132 in IDH1 or the corresponding Arg172 in IDH2, and Arg140 in IDH2. These three residues are located in the enzyme’s active sites, suggesting direct impact of the mutations on the catalytic properties of the enzymes [7]. Because IDH1 and IDH2 mutations affect the enzymatic active site, where isocitrate and NADPH bind, they result in aberrant IDH proteins with neomorphic enzymatic activity. This leads to a product shift of the catalytic reaction from a-KG reduced to the R-enantiomer of 2-hydroxylglutarate (R-2-HG) [11]. As R-2-HG is structurally similar to a-KG, it acts as a competitive inhibitor of a-KG [12] and inhibits a-KG-dependent dioxygenases that are involved in various cellular processes including histone demethylation [13], DNA modification, and adaptation to hypoxia [5]. In addition, IDH mutations can modify the bone marrow microenvironment via paracrine R-2-HG activity [14]. Secretion of R-2-HG by leukemic cells induces NF-kB stabilization and transcriptional activation in stromal cells, which in turn induces IL-6, IL-8 and C5 secretion from stromal cells that stimulates AML cell proliferation.
Second, IDH1/2 mutations occur in a mutually exclusive manner in most cases, and in a highly restricted tumor spectrum.
Finally, IDH mutations generally represent early genomic events in disease development and progression, thus they are present in the dominant clone and remain relatively stable throughout the disease course [15, 16]. As a matter of fact, mutations in IDH genes, especially in IDH2, are occasionally recognized as part of age-related clonal hematopoiesis of indeterminate potential [17] and clonal cytopenia of undetermined significance (CCUS). Malcovati et al. [18] reported that the presence of IDH mutations is a risk factor for developing myeloid malignancy when present in association with TET2, ASXL1 or DNMT3A as part of CCUS. Taken together, these biological features render IDH1/2 mutations reliable and specific biomarkers.
IDH mutations in Korean AML patients
To investigate the distribution and characteristics of IDH1/2 mutations in Korean AML patients, 236 bone marrow samples were collected from three tertiary centers in Korea (Chonnam National University Hwasun Hospital, Samsung Medical Center and Seoul National University Hospital). DNA extraction was performed using Qiagen kit.
We performed targeted resequencing with a customized design: TruSeq Custom Amplicon (Illumina, San Diego, CA) using the MiSeq sequencing platform (Illumina). TruSeq Custom Amplicon is a fully integrated end-to-end amplicon sequencing solution, including online probe design, assay, and sequencing. Online probe design was performed by entering probes into the Design Studio software (Illumina), arbitrarily selected from the medical literature, on the basis of the presence of described mutations with an established role in response to targeted therapies and/or in current treatment paradigms. Once the design was completed, TruSeq Custom Amplicon kit produced the required targeted amplicons with the necessary adapters and indices for sequencing on the MiSeq system without any additional processing. Library preparation and sequencing have been performed according to the manufacturer’s procedures. Quantified libraries were sequenced using the 2×150 bp configuration (300 cycles) and ran on a V2 sequencing flow cell. After sequencing reads were produced, raw de-multiplexed reads from the MiSeq sequencer were aligned to the reference human genome (UCSC build hg19) using the Burrows-Wheeler Aligner (BWA) [19], running in paired-end mode. To ensure good call quality and to reduce the number of false positives, samples underwent Base Quality Score Recalibration (BQSR), using the Genome Analysis Toolkit (GATK) [20]. To detect putative somatic mutations and FLT-ITD, Mutect2 of GATK and ITDetect (version 1.4, self-detection tool in the laboratory) were used. Variant calls were annotated with biological information using AnnoVar and VEP [21, 22]. Mutations were annotated with the 1000 Genomes project, ClinVar, and Catalogue of Somatic Mutations in Cancer (COSMIC, version 92) [23-25]. Variant allelic fraction was determined between 25% and 50%, to exclude non-somatic cells and indels, which are found in lower variant allele frequency and considered as false positive due to sequencing. Additionally, variants with depth lower than 10 were excluded. To retrieve variants that matched targeted regions and genes, and were clinically important, variants that did not map to exons, were not listed in ClinVar, and were splicing or benign variants, were excluded. For detailed analysis and visualization of variant information, ggplot2 and comat of R package were used [26-28].
We found that IDH1/2 mutations were present in 98/236 (41.53%) of the patients (Fig. 1A). To compare our samples with other published AML samples, we obtained oncoplot (Fig. 1B) and mutation profiling data from 822 samples from cBioPortal (622 from OHSU and 200 from TCGA) [2, 29]. We noted that the generally accepted order of mutation frequency in AML was also observed in Korean AML patients, with FLT3-ITD being the most common mutation followed by NPM1.
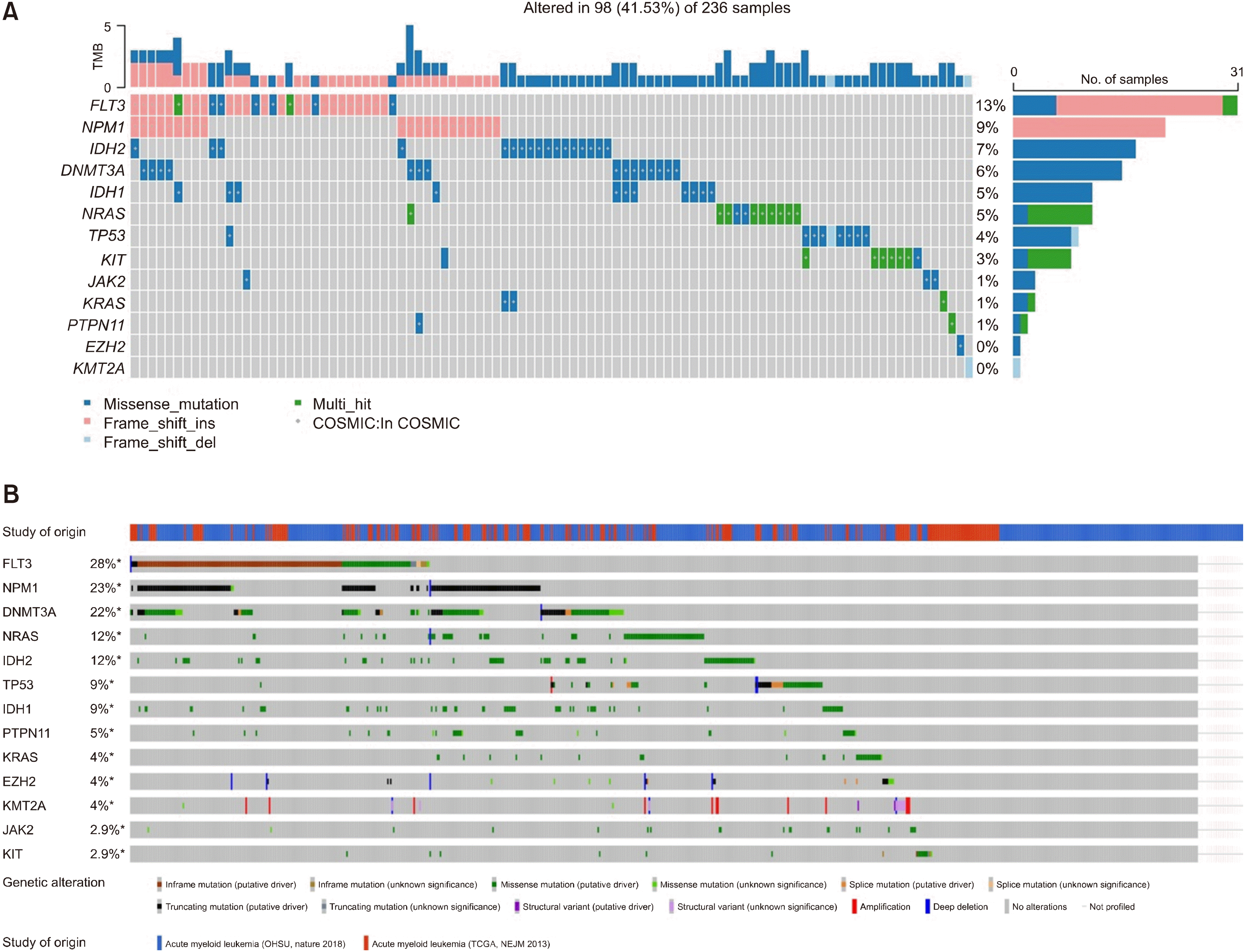 | Fig. 1
(A) Oncoprint with mutation-profiled Korean acute myeloid leukemia samples. Blue: missense mutation, pink: frameshift insertion, sky blue: frameshift deletion, and green: multi-hit. Mutations listed in the COSMIC database are marked with grey dots. (B) Oncoprint obtained from cBioPortal. Clinical and genomic data (622 from OHSU and 200 from TCGA) were merged.
|
Association with other mutations
The association between IDH1/2 mutations and NPM1 or DNMT3A mutations in AML is relatively well-known [2, 3]. More specifically, IDH1 and IDH2 R140 are usually found in combination with NPM1 mutations, while IDH2 R172 is mutually exclusive with NPM1 mutations. This finding, in combination with the observation that more severe aberrations in metabolic activity and higher R-2-HG levels are observed in IDH2 R172, suggests distinct clonal ancestry and biological implications. In addition, IDH1 and IDH2 mutations co-occurred with FLT3-ITD in 15–27% and 8–30% of AML patients, respectively [30].
Co-occurrence or mutual exclusive interactions between genes were also examined in Korean patients (Fig. 2A). IDH1 and IDH2 mutations were found to be mutually exclusive. Overall, IDH1 mutations showed strong association with DNMT3A mutations, and IDH2 mutations with KRAS mutations. These findings are in line with publicly available data (Fig. 2B), which showed close relationship between mutations in IDH1 and DNMT3A [odds ratio (OR)= 6.5490406, P=0.02531], and in IDH1 and NPM1 (OR= 2.397071, P=0.2547) [31]. Interestingly, IDH2 mutations tended to occur together with FLT3 and NPM1 mutations. Additionally, IDH2 mutations were significantly associated with KRAS mutations (OR=27.90556, P=0.01408), which was not observed in public data. Whether this is a true finding or a skewed finding resulting from the limited number of patients requires further investigation.
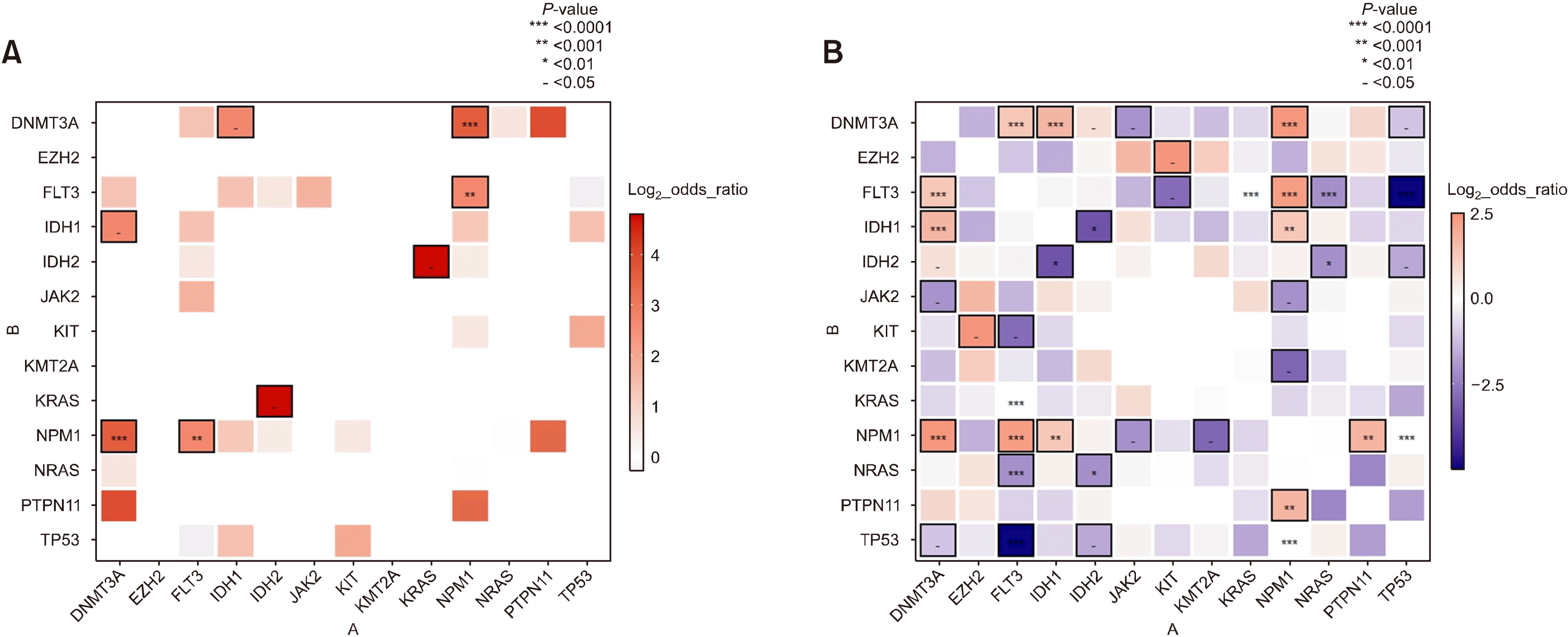 | Fig. 2
(A) Co-occurrence plot displaying interactions between genes in Korean patients. Red indicates higher degree of co-occurrence between two genes, while blue indicates higher degree of mutual exclusiveness. P-values from each Fisher’s exact test are grouped into five groups, and the groups with P>0.05 are not displayed. The remaining four groups with P<0.05 are displayed on a scale for each section. In the case that the absolute value of log2 odds ratio was at least 1 and the P-value is marked, the box’s margin is marked black. When mutations of two genes did not occur in one sample, the odds ratio could not be calculated, therefore, many mutual exclusive interactions identified in Fig. 1A are not shown in this plot. (B) Co-occurrence plot using publicly available data matching with Fig. 1B. The number of patients is larger than the Korean cohort. Accordingly, a higher number of mutually exclusive signals were detected in the public database than in the Korean cohort.
|
In Korean patients with IDH1/2 mutations (Fig. 3A), co-occurrence with FLT3-ITD mutations was identified at a rate of 27% with IDH1 (FLT3-ITD/IDH1: 3/11), and 6.6% with IDH2 (FLT3-ITD/IDH2: 1/15). Specific mutational hotspots were also examined. IDH1 R132 and IDH2 R140 occurred together with NPM1 mutations, while IDH2 R172 showed a mutual exclusive relationship with NPM1 mutations (OR=23.69956, P=0.009588) (Fig. 3B). Similarly, and as expected, in publicly available data (Fig. 3C, D) the co-occurrence between NPM1 mutations and IDH1 R132 (OR=2.585159, P=0.0003675), and that between NPM1 mutations and IDH2 R140 (OR=1.738254, P=0.044) were well-noted. Moreover, mutual exclusiveness between IDH2 R172 and NPM1 mutations (OR=0, P=0.03025) was also observed. On the other hand, in the Korean AML cohort, missense mutations in FLT3-ITD were identified to co-occur with IDH2 R172 (OR=23.69956, P=0.009588). However, in the public data, IDH2 R172 showed mutual exclusiveness with FLT3-ITD mutations (OR=0.16318, P=0.05037). This latter finding requires further investigation.
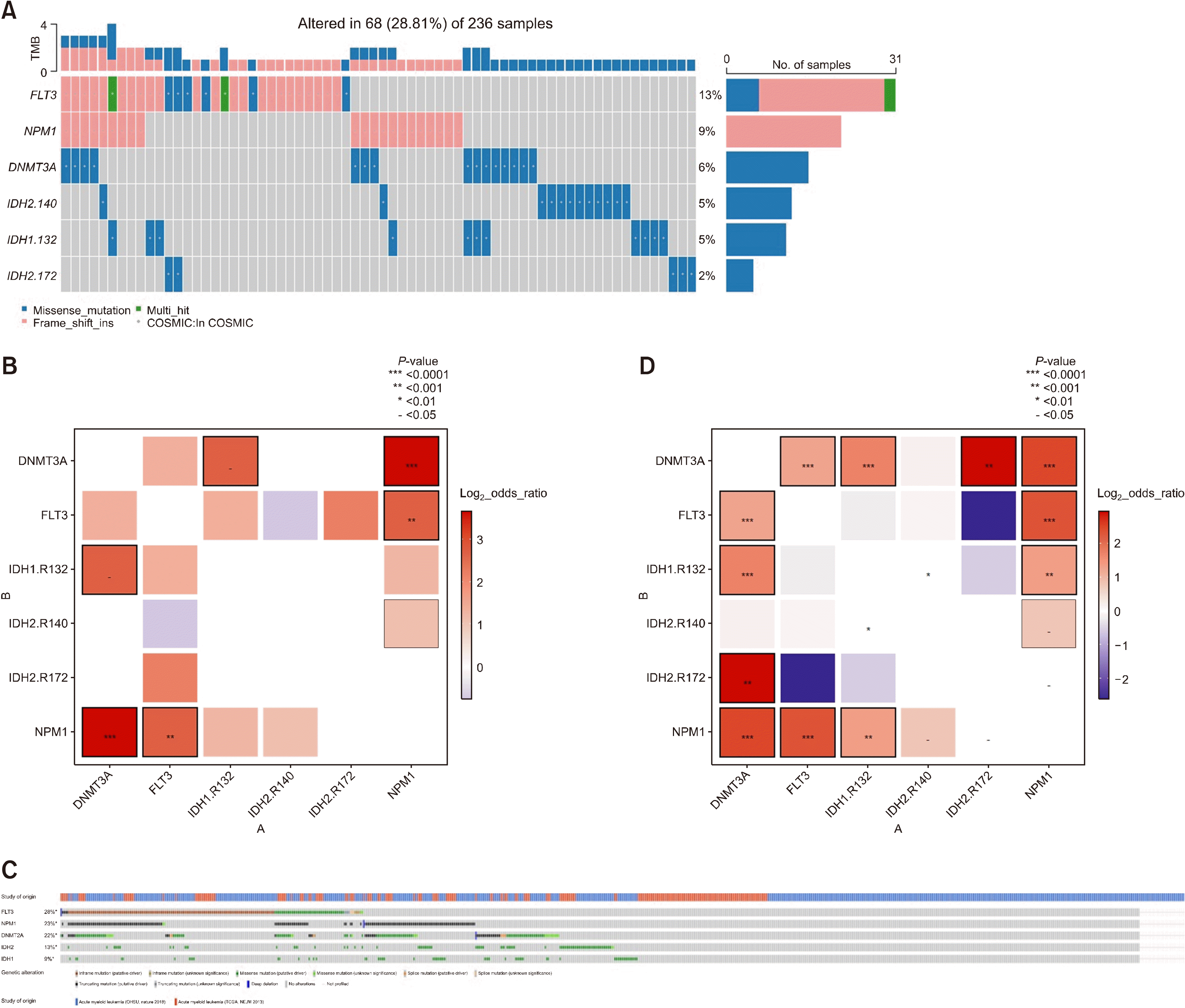 | Fig. 3
(A) Oncoplot showing Korean patients with FLT3, NPM1, DNMT3A, IDH1 and IDH2 mutations. Hotspots are shown for IDH1 and IDH2. (B) Interactions between five genes from Korean AML patients shown in the same way as previously displayed. (C) Oncoplots using public data from cBioPortal regarding five genes. (D) Interactions between five genes from samples from cBioPortal shown in the same way as previously displayed. In cases of white color boxes and P-value marking, odds ratio is zero because there are no events of mutations in both genes.
|
Prognosis
The prognostic impact of IDH1/2 mutations in AML remains controversial. In their recent meta-analysis of 12,747 patients from 33 studies, Xu et al. [32] showed that IDH1 mutations were associated with worse survival and lower likelihood of complete remission (CR), especially in patients with NK. On the other hand, both IDH2 R140 and R172 mutations were associated with better survival outcomes, especially for intermediate-risk groups. However, interpretation of the results requires scrutiny, as there were differences between patient cohorts, variations between study methodologies, and grouping of mutations for prognostic analysis ranged widely. Of note, Molenaar et al. [15] showed that ancestral IDH1 mutations were associated with worse prognosis compared with subclonal IDH1 mutations. Such clonal hierarchy had no survival impact for patients with IDH2 mutations.
Treatment
Ivosidenib (IDH1 inhibitor): Ivosidenib (Tibsovo) is an orally administered IDH1 inhibitor that reduces the total serum R-2-HG level and induces cell differentiation. The recommended dose is 500 mg orally once daily, until disease progression or occurrence of unacceptable toxicity.
The approval of ivosidenib by the US-FDA in 2018 was based on results from ivosidenib monotherapy in adult relapsed/refractory (R/R) AML patients with IDH1 mutations (NCT02074839). The overall response rate (ORR) was 41.6% with a median response time of 6.5 months, and the CR rate was 21.6% with a median duration time of 9.3 months. Adverse events occurred in 99% of the patients, most common being diarrhea (31%), leukocytosis (30%), and febrile neutropenia (29%). There were no deaths related to adverse events, although grade ≥3 adverse events occurred in 21% of the patients. The most common treatment-related grade ≥3 adverse events were prolonged QT interval, differentiation syndrome and leukocytosis, which were also the most common reasons for drug withdrawal. Differentiation syndrome occurred in approximately 11% of the patients with a median onset time of 29 days [35, 36].
Enasidenib (IDH2 inhibitor): Enasidenib (Idhifa) is an oral specific allosteric inhibitor of IDH2 that shows more robust inhibitory effects on IDH2 R172 than on IDH1 R140 mutations, although it has inhibitory effects on both mutations [37]. Enasidenib has been shown to reduce total serum R-2-HG level by more than 90%, to reverse abnormal epigenetic changes, and to induce differentiation of mutant IDH2 AML cells both in vitro and in murine xenograft models. The recommended oral dose is 100 mg once daily for at least 6 months or until disease progression or intolerable adverse reactions.
The approval of ivosidenib by the US-FDA in 2017 was based on the results of phase 1/2 trial (NCT01915498) in adult R/R AML patients. ORR was 40.3% (CR, 19.3%) with a median OS of 9.3 months. Treatment-related adverse events included mainly elevated bilirubin (81%), nausea (50%), and diarrhea (43%). Differentiation syndrome of any grade occurred in 14% of the patients (grade ≥3 occurred in 7% of patients), and all were manageable with glucocorticoids and diuretics [38, 39].
Resistance: Mutations in NRAS and MAPK pathways have been proposed as causes of primary resistance. Due to increased reactive oxygen species (ROS) levels, genes associated with MAPK signaling are highly enriched in IDH1 mutant cells and transcription of cyclin-dependent kinase (CDK) inhibitors are repressed, leading to enhanced cell proliferation [40]. Moreover, due to inhibition of specific a-KG-dependent dioxygenases by R-2HG, IDH mutations can induce a “BRCAness” phenotype characterized by a homologous recombination defect leading to increased double-strand DNA damage that renders tumor cells sensitive to PARP inhibition [41].
The mechanisms of acquired resistance remain uncertain, but the second site mutation of the allele and the mutual conversion of IDH mutant subtypes are the two mains mechanisms.
Go to : 
CONCLUSION
IDH1/2 mutations are becoming increasingly important for AML treatment, thus they should be examined and followed-up throughout the clinical course of the disease. Future clinical trials should focus on determining the best patient settings and optimizing the sequential application of effective agents to further improve clinical outcomes using tailored treatment in AML.
Go to :
 XML Download
XML Download