

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hepatocellular carcinoma (HCC), which represents about 75% to 85% of primary liver cancers, is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer-related deaths worldwide. Most HCCs develop in patients with liver cirrhosis, with hepatitis B virus (HBV) infection being the leading cause of liver cancer and liver cancer deaths globally (33%) [1]. Although multiple genes and molecular pathways were shown to be involved in the development of HCC, the exact mechanisms underlying HCC tumorigenesis and progression remain unclear. Identification of key genes and molecular mechanisms underlying the development of HCC is important for determining future diagnostic biomarkers and therapeutic targets.
High-throughput sequencing methods such as microarray analysis can enable the profiling of large gene expression datasets for various diseases including HCC. Thus, it may provide clues for understanding the mechanism of pathogenesis including carcinogenesis. Combinations of bioinformatics and microarray technology have identified various key genes essential for the development of HCC [2,3].
The present study analyzed the GSE121248 gene dataset of 70 HCCs and 37 adjacent normal liver tissue samples from patients with HBV-associated HCC obtained from the Gene Expression Omnibus (GEO) database. This dataset was investigated to identify differentially expressed genes (DEGs) in HCCs and adjacent non-cancerous liver tissue samples. Using bioinformatics methods, key genes and pathways were identified using a series of functional analyses.
Go to : 
MATERIALS AND METHODS
Microarray data
The GSE121248 gene dataset based on the GLP570 platform was downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). This dataset included RNA array data for 107 tissue samples obtained from patients with HBV-associated HCC, including 70 HCCs and 37 adjacent normal liver tissue samples.
Identification of differentially expressed genes
Data normalization and differential expression analyses were performed using R packages affy and limma (R Foundation for Statistical Computing, Vienna, Austria) and the GEO2R online tool (http://www.ncbi.nlm.nih.gov/geo/geo2r/). Probe IDs without matches or multiple gene matches were removed. Adjusted p-values were applied to correct false-positive results using the Benjamini and Hochberg false discovery rate method. Adjusted p-values < 0.05 and absolute log2 FC values > 2 were considered cut-off criteria for defining DEGs.
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genome (KEGG) pathway enrichment analysis
To explore the biological significance of DEGs, the Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.ncifcrf.gov) and R packages clusterProfiler were used for visualization and enrichment analyses, including analyses of biological processes, cellular components, molecular functions, and KEGG pathways. A p-value < 0.05 was considered statistically significant.
Integration of protein-protein interaction network
To determine connections between proteins encoded by DEGs and significant gene modules involved in the development of HCC in HBV-infected patients, protein-protein interaction (PPI) network analysis was performed using the Search Tool for the Retrieval of Interacting Genes (STRING) database (http://www.string-db.org/). Cytoscape (3.8.0) was applied to construct a PPI network with a confidence score ≥ 0.4. Modules were analyzed with Cytoscape plugins Molecular Complex Detection (MCODE) and GOClue. A p-value < 0.05 was considered statistically significant.
Validation of hub genes
To ensure the credibility of microarray analysis of GSE121248, expression levels of mRNAs encoded by 14 hub genes were compared in the GSE121248 and The Cancer Genome Atlas (TCGA) databases. The Gene Expression Profiling Interactive Analysis (GEPIA; http://gepia.cancer-pku.cn/index.html), a web server for analyzing the expression of mRNA sequences in tumor and normal tissue samples from the TCGA and Genotype-Tissue Expression (GTEx) projects, was utilized to construct boxplots to visualize the expression of mRNAs encoded by hub DEGs in HCC and normal liver tissue samples based on the TCGA database.
Overall patient survival
The GEPIA database was utilized to construct Kaplan–Meier curves for the overall survival of patients with HCC as a function of the expression of the 14 hub genes. The median value of gene expression was used as the cutoff value for grouping. Curves were compared by log-rank tests. Hazard ratios and 95% confidence intervals were calculated. A p-value < 0.05 was considered statistically significant. The study protocol was approved by the Institutional Review Board of Asan Medical Center (IRB No. 2021-0683), which waived the requirement for informed consent due the use of publicly open data.
Go to :
RESULTS
Identification of DEGs
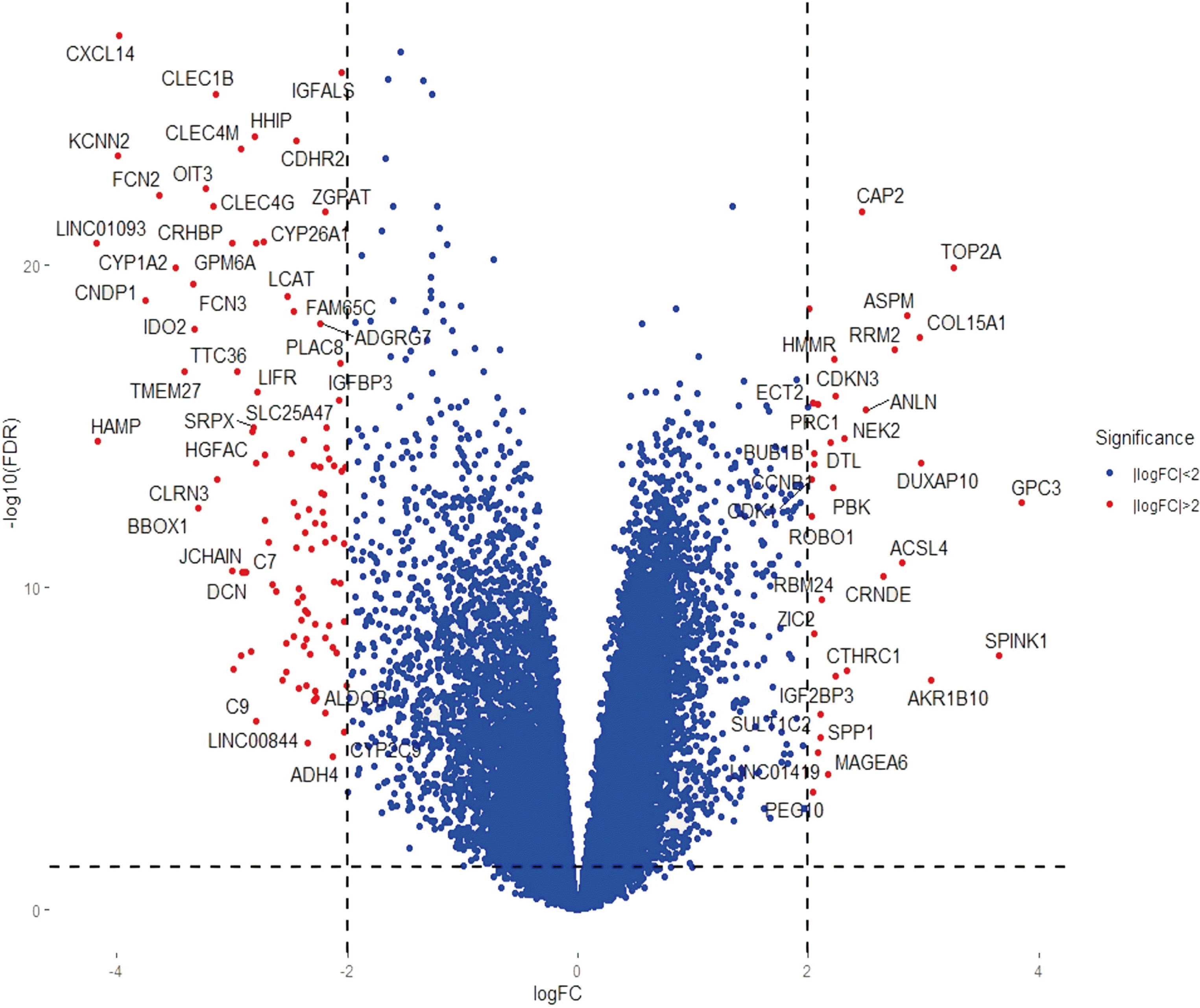
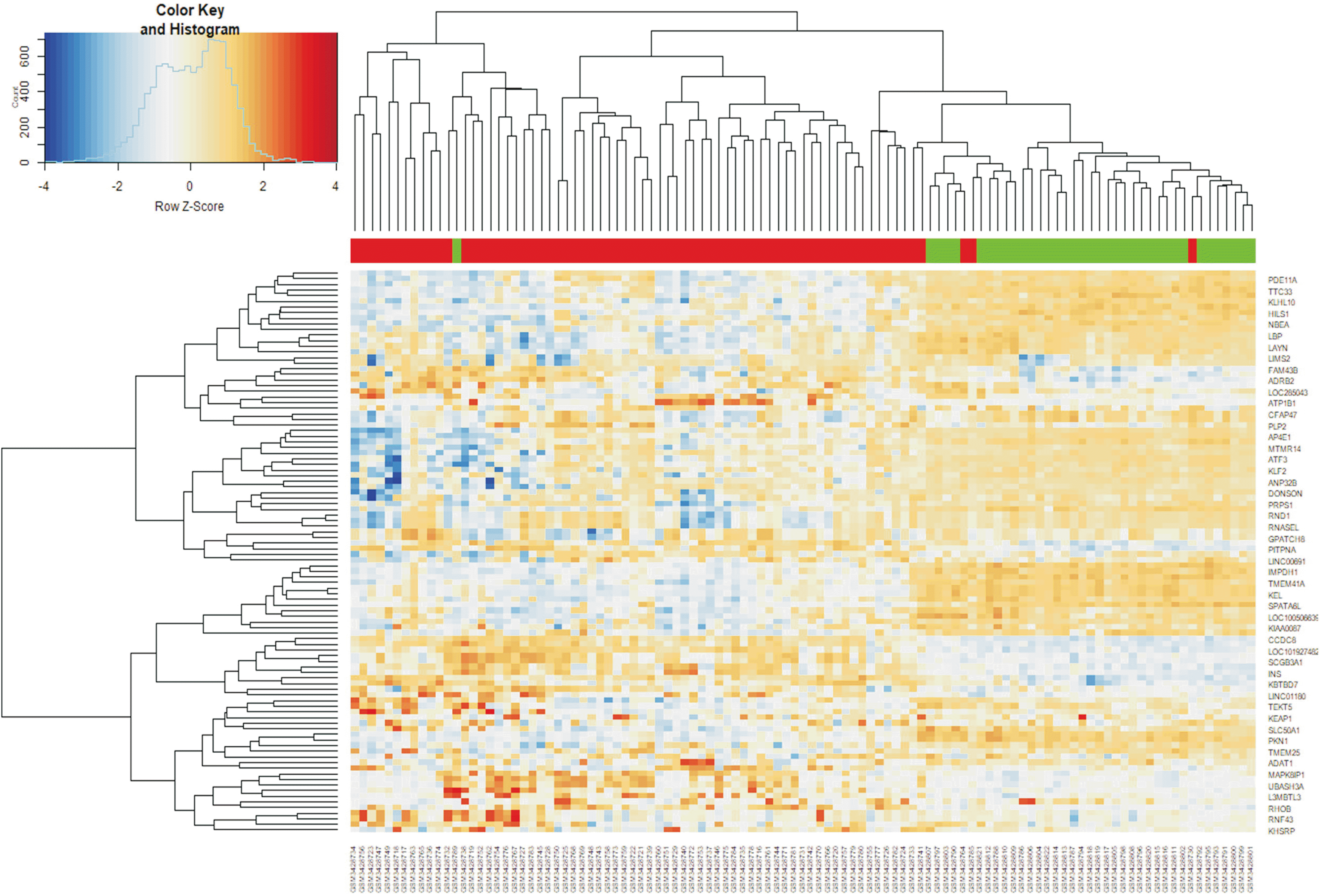
The GSE121248 dataset, which included 70 HCC tissue samples and 37 adjacent normal tissue samples from patients with HBV-associated HCC, was analyzed using the R packages affy and limma and the GEO2R online tool to identify DEGs with thresholds of | log2 FC | > 2 and adjusted p-values < 0.05. Of 134 DEGs identified, 34 were significantly higher and 100 significantly lower in HCC than in adjacent normal liver tissues. Fig. 1 is a volcano plot showing the distribution of identified DGEs and Fig. 2 is a heatmap showing expression levels of the 100 top DEGs with | log2 FC | > 2.
GO and KEGG pathway enrichment analysis
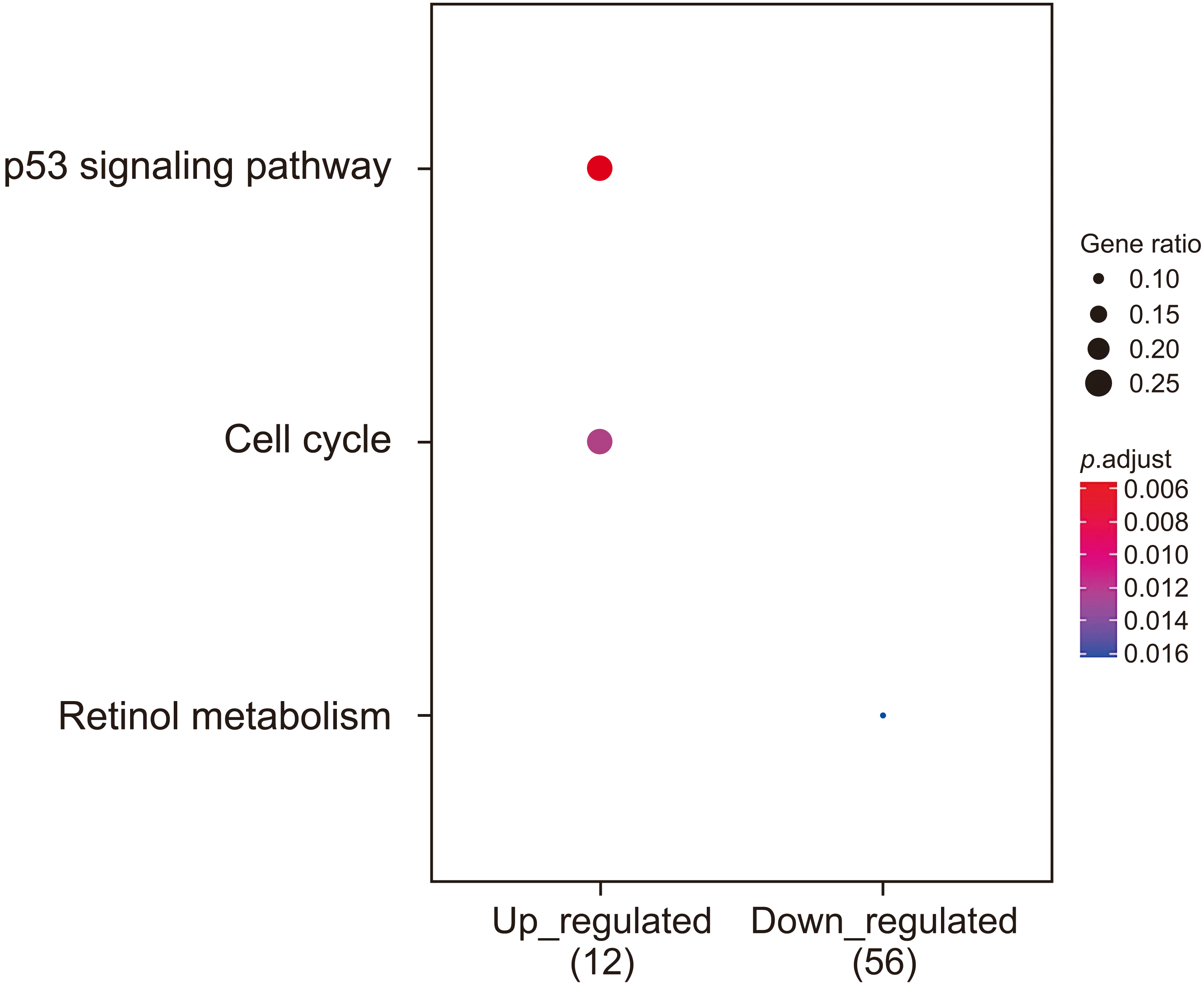
To determine the function of the identified DEGs, functional and pathway enrichment analyses were performed using DAVID and R packages clusterProfiler. GO enrichment analysis for assessing biological processes revealed that the 34 up-regulated DEGs were mainly involved in nuclear division and organelle fission (Fig. 3), whereas the 100 down-regulated DEGs were mainly involved in steroid and hormone metabolism (Fig. 4). Analysis of cellular components showed that up-regulated DEGs mainly participated in spindle and midbody formation (Fig. 3), whereas down-regulated DEGs mainly participated in the synthesis of collagen-containing extracellular matrices and blood microparticles (Fig. 4). Analysis of molecular function showed that up-regulated DEGs were enriched in histone kinase activity (Fig. 3), whereas down-regulated DEGs were mainly enriched in oxidoreductase activity and activities related to paired donors, including the incorporation or reduction of molecular oxygen and monooxygenase activity (Fig. 4). KEGG pathway analysis found that up-regulated DEGs were mainly involved in the p53 signaling pathway and the cell cycle, whereas down-regulated DEGs were mainly involved in retinol metabolism (Fig. 5).
 | Fig. 3Gene ontology enrichment analysis of the 34 up-regulated differentially expressed genes with log2 FC > 2. FC, fold change; BP, biological process; CC, cellular component; MF, molecular function.
|
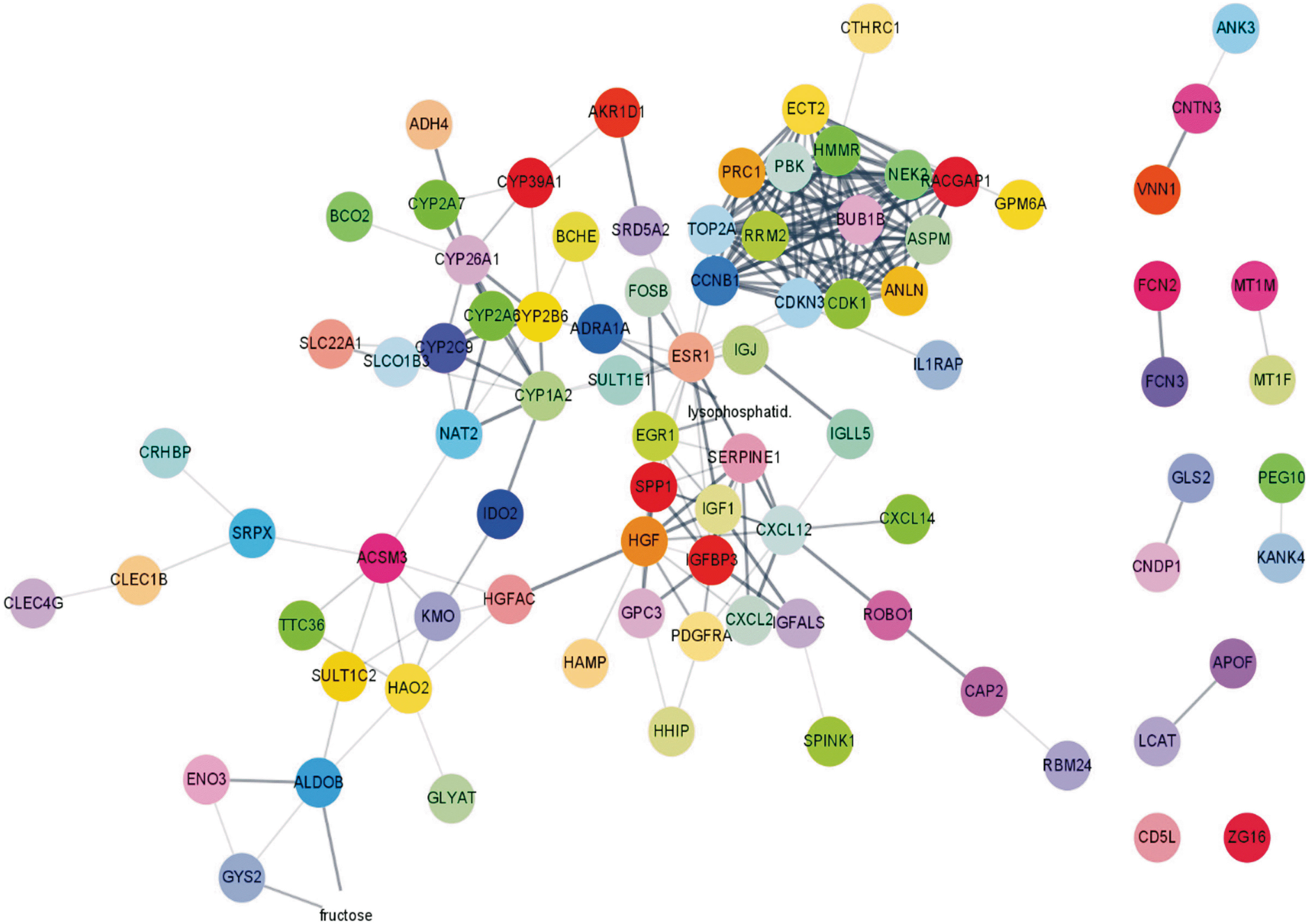
Integration of the protein-protein interaction network
As constructed by Cytoscape (3.8.0), based on the STRING online tool, the PPI network of the 134 identified DEGs was found to consist of 134 nodes and 370 edges, with an average node degree of 5.82 and an average local clustering coefficient of 0.525 (Fig. 6). Based on the degree of connectivity of these 134 DEGs, the Cytoscape plug-in MCODE was able to create a significant module that included 14 nodes and 91 edges (Fig. 7). These 14 nodes represent 14 hub genes of high connectivity, including ANLN, ASPM, BUB1B, CCNB1, CDK1, CDKN3, ECT2, HMMR, NEK2, PBK, PRC1, RACGAP1, RRM2, and TOP2A. Subsequent enrichment analysis showed that these 14 hub genes were mainly associated with nuclear division, organelle fission, spindle and midbody formation, protein serine/threonine kinase activity, histone kinase activity, the p53 signaling pathway, and the cell cycle (Fig. 8, Table 1).
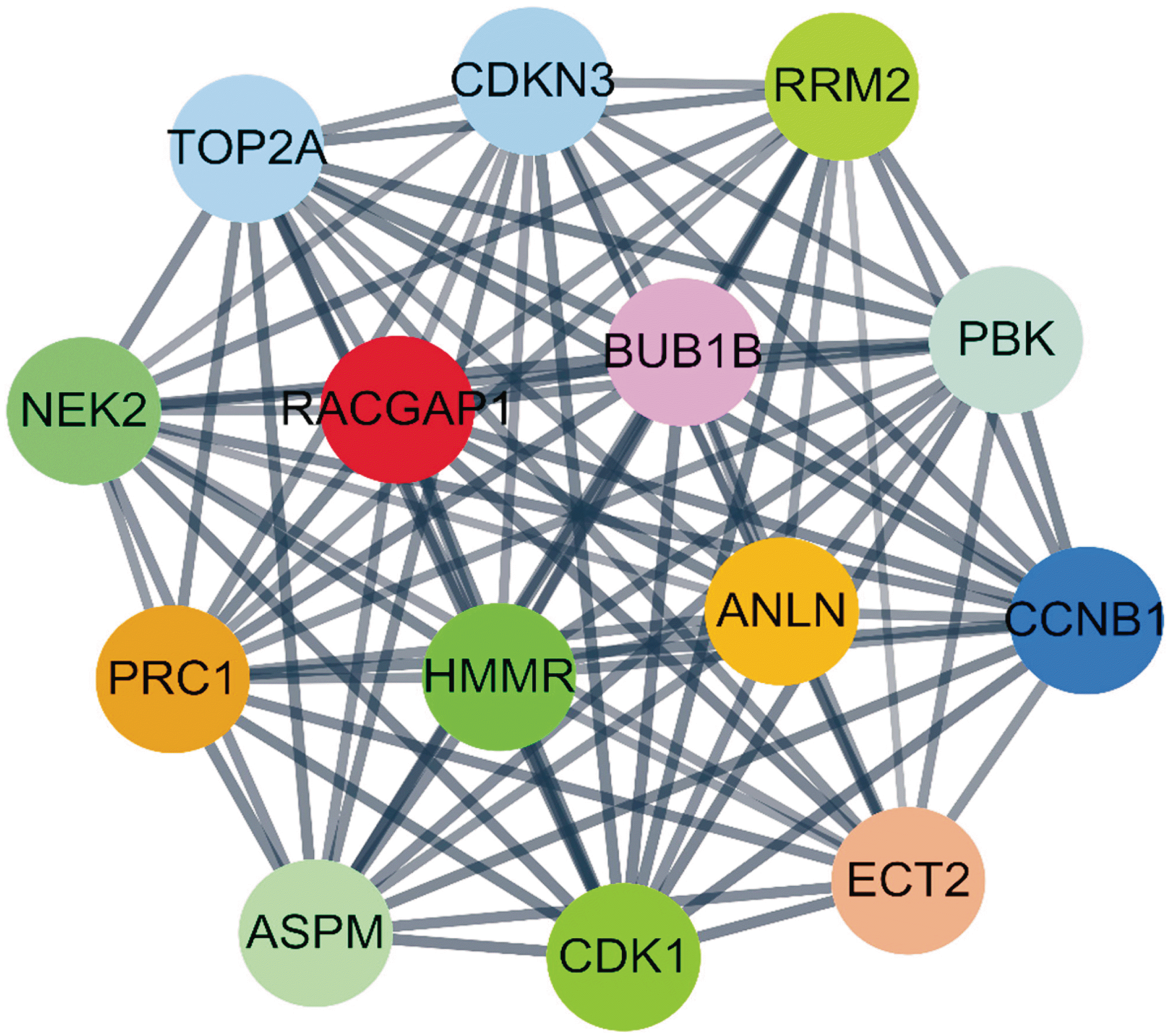 | Fig. 7Protein-protein interaction network of the top 14 hub genes with a high degree of connectivity.
|
 | Fig. 8Enrichment analyses of the 14 hub genes in the most significantly enriched module. Results of gene ontology (A) and Kyoto Encyclopedia of Genes and Genome (B) enrichment analyses are shown.
|
Table 1
Pathway enrichment analysis of the 14 hub genes in the most significant modules
![]()
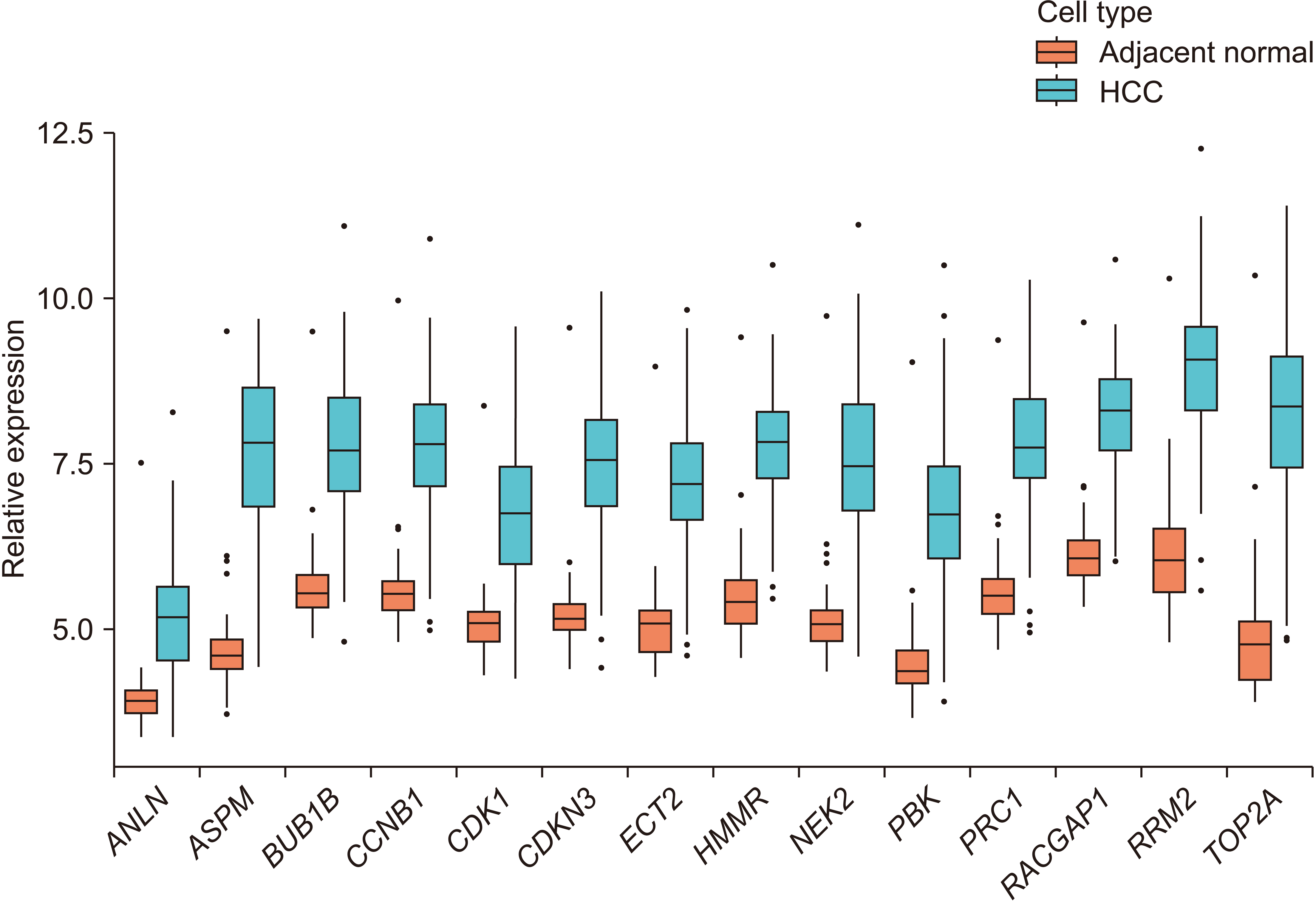
Validation of hub genes by the TCGA database
Analysis of expression levels of mRNAs encoded by the 14 hub genes in the GSE121248 dataset showed that all 14 were expressed at higher levels in the 70 HCC samples than in the 34 adjacent normal tissue samples (Fig. 9). To validate these findings, mRNA expression levels of the 14 hub genes in HCC and normal samples in the TCGA database and GEPIA database were determined (Fig. 10). Similar to findings in the GSE121248 dataset, levels of mRNAs encoded by all 14 hub genes were significantly higher in HCC than in normal liver tissues in the GEPIA database.
Overall patient survival
Kaplan–Meier curves obtained from GEPIA (Fig. 11) showed close associations between expression levels of the 14 hub genes and the overall survival of HCC patients. High expression levels of 12 hub genes (all 14 hub genes except CDKN3 and RACGAP1) were strongly correlated with poor patient prognosis (p < 0.05).
Go to :
DISCUSSION
HCC is currently the most common type of primary liver cancer in adults. It is also the most frequent cause of death in patients with liver cirrhosis. HCC in many countries, including Korea, is closely related to HBV infection. Although patients infected with HBV are regularly screened for HCC by measuring blood levels of alpha-fetoprotein (AFP) and abdominal ultrasonography every six months, HBV-associated HCC remains the main cause of mortality in HBV-infected patients [4]. The identification of new biomarkers and regulatory pathways related to the development of HCC in HBV-infected patients can enhance early detection, diagnosis, and treatment of HCC. The present study utilized bioinformatics to analyze the GSE121248 gene expression dataset and screen for key genes and molecular pathways that are closely related to the development of HCC. Using cut-off criteria of p < 0.05 and | log2 FC | > 2, this analysis identified 134 DEGs, of which 100 were down-regulated and 34 were up-regulated in HCC than in normal liver tissues. To obtain a comprehensive understanding of these DEGs, we performed bioinformatics analyses, including GO enrichment, KEGG pathway, and PPI network analyses. To validate the 14 hub genes identified, we also analyzed their expression levels using the GEPIA website and assessed their effects on overall patient survival using the TCGA database.
GO enrichment analysis revealed that the 34 up-regulated DEGs were significantly enriched in genes involved in nuclear division and organelle fission, whereas the 100 down-regulated DEGs were highly enriched in genes involved in steroid metabolism. KEGG pathway analysis showed that the 34 up-regulated genes were enriched in genes involved in the p53 signaling pathway, whereas the 100 down-regulated genes were significantly enriched in genes involved in retinol metabolism. PPI network analysis was performed using STRING. Hub genes were identified by MCODE, a Cytoscape plug-in. The 14 hub genes identified by PPI network analysis included ANLN, ASPM, BUB1B, CCNB1, CDK1, CDKN3, ECT2, HMMR, NEK2, PBK, PRC1, RACGAP1, RRM2, and TOP2A. Analysis using the GEPIA website showed that expression levels of all 14 mRNAs were much higher in HCC than in normal tissue samples. Survival analysis using the TCGA database showed that expression levels of these genes were significantly associated with patient survival.
Anillin (ANLN) is a conserved protein implicated in cytoskeletal dynamics during cytokinesis and cellularization. ANLN is essential for cell division. Therefore, it is critical for development and homeostasis in metazoans. ANLN expression levels are correlated with the metastatic potential of human tumors. For example, colorectal cancer cell lines expressing higher levels of ANLN replicated faster, were more invasive, and had greater levels of migration than cell lines expressing lower levels of ANLN [5]. Overexpression of ANLN may enhance cell migration by cytoskeletal remodeling, promoting the proliferation, invasion, and motility of tumor cells [6]. In addition, overexpression of ANLN involving copy number variation, as well as alterations in DNA and histone methylation, was found to be related to poor prognosis in patients with HCC [7].
The abnormal spindle-like microcephaly-associated (ASPM) protein plays a significant role in neurogenesis and cell proliferation. In addition, ASPM overexpression has been associated with poor outcomes of patients with various cancers [8,9]. ASPM overexpression may predict HCC invasiveness or metastatic potential. It is associated with a higher risk of early tumor recurrence regardless of p53 mutation status or tumor stage [10].
The mitotic checkpoint serine/threonine-protein kinase BUB1 beta (BUB1B) gene encodes a kinase involved in spindle checkpoint function and chromosome segregation. Impaired spindle checkpoint function is involved in the initiation and development of tumors. In addition, BUB1B can promote HCC progression by activating the mTORC1 signaling pathway [11].
Cyclin B1 (CCNB1) is a regulatory protein involved in mitosis. A high level of CCNB1 expression may be associated with the extent of tumor progression and invasion, suggesting that the expression level of CCNB1 might be prognostic of survival outcomes in cancer patients. Transcriptional activation of CCNB1 is essential for the proliferation of HCC. It is closely associated with poor prognosis in patients with HCC [12].
Cyclin-dependent kinase 1 (CDK1), a member of the Ser/Thr protein kinase family, plays an important role in G1/S and G2/M phase transitions of eukaryotic cell cycle by interacting with CCNB1. CDK1 was found to play an essential role in HCC development by regulating subcellular localization of apoptin [13]. Metformin can significantly inhibit HCC cell proliferation by inducing G2/M arrest and reducing the expression of CDK1, suggesting that CDK1 might be involved in the process of HCC cell proliferation [14]. In addition, overexpression of CDK1 in HCC is significantly associated with large tumor size, advanced tumor stage, portal invasion, intrahepatic metastasis, poor differentiation, high AFP level, high Ki-67 index, and poor patient prognosis [15].
Cyclin-dependent kinase inhibitor 3 (CDKN3), which inhibits the activation of cyclin-dependent kinase 2 (CDK2), can promote tumorigenesis in various types of cancer [16]. In contrast to our findings, a recent study has found that CDKN3 is negatively associated with pathological tumor stage and that its inhibition can enhance HCC cell survival [17].
The epithelial cell transformation sequence 2 (ECT2) protein is a Rho-specific exchange factor. Activation of the ECT2/Rho pathway can enhance the progression of several tumors. ECT2 overexpression may promote the progression and early recurrence of HCC by regulating the Rho/ERK signaling axis [18]. High levels of ECT2 have been implicated in other types of cancer [19].
Hyaluronan-mediated motility receptor (HMMR or RHAMM) was originally identified as a soluble protein that altered migratory cell behavior and bound to hyaluronan. HMMR expression is associated with breast cancer risk and the progression of several tumor types. A recent bioinformatics study has found that overexpression of HMMR is positively correlated with HCC tumor grade, pathological stage, and tumor stage [20].
The serine/threonine-protein kinase Nek2 (NEK2) is a microtubule-associated protein that regulates spindle assembly in human cells. It is overexpressed in malignancies. NEK2 upregulation has been observed in primary HCC tissue and HCC cell lines. It is significantly associated with cancer progression, drug resistance, and poor patient prognosis [21]. In contrast to these findings, another study has reported that low expression of NEK2 is associated with HCC progression and poor patient prognosis [22].
Lymphokine-activated killer T-cell-originated protein kinase (PBK) is a serine/threonine-protein kinase associated with the dual specific mitogen-activated protein kinase kinase (MAPKK) family. PBK is involved in the activation of lymphoid cells, which support testicular function and spermatogenesis. Overexpression of PBK has been associated with tumor progression. In HCC, PBK overexpression can promote metastasis through activation of the ETV4-uPAR signaling pathway [23].
Protein regulator of cytokinesis 1 (PRC1) is a protein that regulates antiparallel microtubule cross-linking, promoting the formation of microtubules that support cell shape and regulate cytokinesis. PRC1 is overexpressed in HCC. Its high expression correlates with early HCC recurrence and chemoresistance [24].
Rac GTPase-activating protein 1 (RACGAP1) belongs to the family of Rho GTPase-activating proteins. It plays a key role in controlling the initiation of cytokinesis, transformation, invasive migration, and metastasis. Overexpression of RacGAP1 protein in colorectal cancer is significantly associated with higher T stage, vessel invasion, and distant metastasis. It is therefore significantly related to poor disease-free and overall survival rates [25]. In HCC, overexpression of RACGAP1 correlates with early recurrence and a high migration rate [26].
Ribonucleotide reductase subunit M2 (RRM2) catalyzes the production of deoxynucleotide triphosphates, which are essential for both replicative and repair DNA synthesis. RRM2 is known to play an active role in tumor progression. Elevated RRM2 levels are correlated with poorer prognosis in patients with several types of cancer [27]. Overexpression of RRM2 in HCC correlates with early recurrence and intrahepatic metastasis. It is therefore associated with poor prognosis [28].
The DNA topoisomerase 2 α (TOP2A) gene located on human chromosome 17 encodes the enzyme DNA topoisomerase IIα. TOP2A expression may be a significant predictor of tumor progression, recurrence, and poor survival rate in various types of cancer [29]. In HCC, high expression of TOP2A has been associated with more advanced clinical stages, lower grades of tumor differentiation, high T stages, and resistance to chemotherapy [30].
Patterns of expression of the 14 hub genes identified in this study were similar in other malignancies. In addition, several in vivo, in vitro, and bioinformatics studies have shown that individual expression level of each of these 14 hub genes is correlated with the development and progression of HCC.
The background purpose of the present study was to identify key genes and pathways of HBV-associated HCC as a preliminary study before actual experimental study using tissue samples obtained from our own patients. It is essential to perform external validation studies focusing on results of the present study.
The present study has limitations. This study included only one public database. It is necessary to validate results of this study with external data sets obtained from patients of our institution and other Korean institutions.
In conclusion, this comprehensive bioinformatics analysis identified candidate genes and pathways that might be involved in the progression of HBV-associated HCC. These findings may provide better understanding of the molecular mechanisms and potential biomarkers of HCC. Although further experiments with additional patients are required to confirm these findings, results of the present study may suggest future genomic treatment targets for patients with HCC.
Go to :
 XML Download
XML Download