

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) is an autosomal recessive inherited disease that disrupts genes encoding protein transporters responsible for bile formation [1-3]. PFIC was first described in an Amish kindred and called Byler disease [4]. Clinical manifestations of PFIC include progressive jaundice, pruritus, and growth retardation. Subsequently, patients develop liver cirrhosis and hepatic failure.
PFIC is classified into three types (type 1–3). PFIC type 1 is caused by a mutation of ATP8B1 gene encoding FIC1 protein [2]. PFIC type 2 is caused by a mutation of ABCB11 gene encoding bile salt export pump (BSEP). PFIC type 3 is caused by a mutation of ABCB4 gene encoding MDR3 protein (class III multidrug resistance p-glycoprotein) [3].
Liver transplantation (LT) has been regarded as the only effective treatment for PFIC [5]. The estimated incidence rate of PFIC is 1 : 50,000 to 1 : 100,000 births [6]. The experience of LT for PFIC in Korea is very limited. Thus, the objective of this study was to investigate the clinicopathological features and posttransplant courses in seven pediatric LT recipients who were diagnosed with PFIC.
Go to : 
PATIENTS AND METHODS
This was a retrospective single-center study on LT series in patients with PFIC. The LT database of our institution was searched to identify pediatric patients who underwent LT for PFIC from January 2013 to June 2020. All patients were followed up until April 2021 or death by reviewing institutional medical records with the assistance of the National Health Insurance Service in Korea. The study protocol was approved by the Institutional Review Board of our institution (No. 2020-0836). The requirement for informed consent was waived due to the retrospective nature of this study. This study was performed in accordance with the ethical guidelines of the World Medical Association Declaration of Helsinki 2013.
Genomic DNA was isolated from peripheral blood leukocytes. Twenty-seven exons with exon-intron boundaries of ATP8B1 and 28 exons with exon-intron boundaries of ABCB11 were amplified by polymerase chain reaction (PCR). The PCR products were electrophoresed and directly sequenced. DNA sequences were compared with GenBank (http://www.ncbi.nlm.nih.gov) reference DNA sequences (NT_0025028.14 and NT_005403.17 for genomic sequences of ATP8B1 and ABCB11, respectively; and NM_005603.4 and NM_003742.2 for ATP8B1 and ABCB11 mRNAs, respectively). Detailed procedures of gene analysis were described previously [7]. BSEP immunohistochemical staining was performed with a BSEP antibody (Santa Cruz Biotechnology, Dallas, TX, USA). Survival curves were estimated using the Kaplan–Meier method. All statistical analyses were performed using IBM SPSS ver. 22 (IBM Corp., Armonk, NY, USA).
Go to :
RESULTS
Pretransplant clinical findings
Clinical characteristics of seven patients with PFIC are presented in Table 1. Two patients (case no. 3 and no. 6) were diagnosed with PFIC type 1. The other five patients were diagnosed with PFIC type 2. Five were females and two were males. Age at PFIC onset was at birth in all seven patients. Five patients were Koreans and two were Arabians. Jaundice was present in all seven patients. Mean pretransplant total and direct bilirubin levels were 16.1 ± 8.1 mg/dL and 12.4 ± 6.2 mg/dL, respectively. Three of four patients showed high elevation of serum alpha-fetoprotein (AFP) levels.
Table 1
Peritransplant clinical profiles of seven patients
F, female; M, male; LT, liver transplantation; PT INR, prothrombin time international normalized ratio; AFP, alpha-fetoprotein; PFIC, progressive familial intrahepatic cholestasis; GRWR, graft-to-recipient weight ratio; NA, not available; DD, deceased donor; HRLLS, hyper-reduced left lateral section; LLS, left lateral section; WL, whole liver.
![]()
Transplantation profiles
The median patient age and body weight at LT were 10 months (range, 3–46 months) and 7 kg (range, 4.1–12.2 kg), respectively. Types of donors were mothers of patients in four and deceased donors in three. Two 3-month-old infant recipients received hyper-reduced left lateral section grafts, one from the mother (case no. 4) and the other one from a deceased donor (case no. 1) [8]. A 10-month-old patient underwent LT with a whole liver graft recovered from an infant donor (case no. 6) [9]. The remaining five patients received left lateral section grafts. Mean graft-to-recipient weight ratio was 3.6% ± 1.1%. Peritransplant imaging findings are individually presented in Fig. 1, matching with the case no. Standardized surgical techniques for pediatric LT were used for implantation of left lateral section grafts.
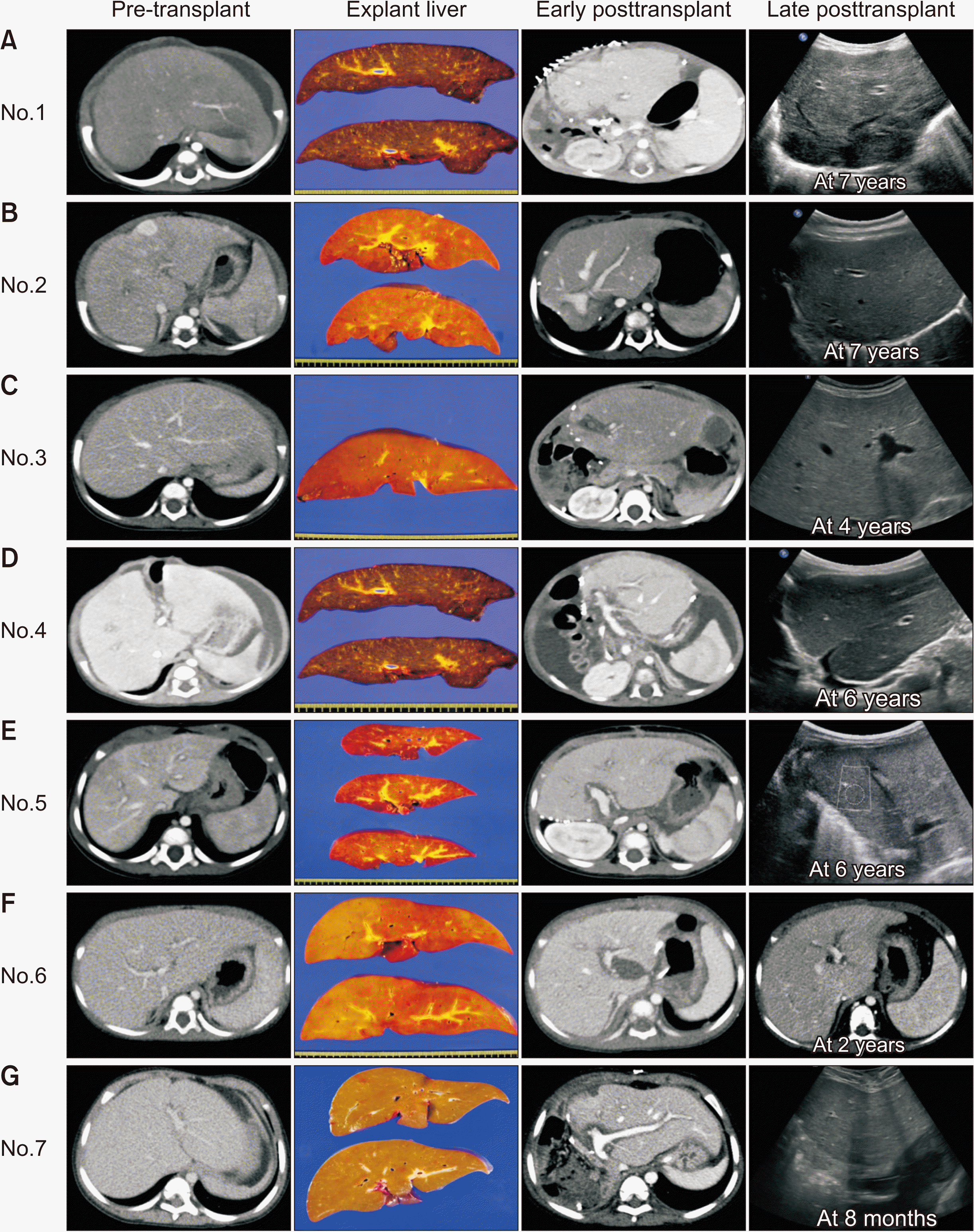 | Fig. 1Peritransplant imaging findings of seven cases with pretransplant, explant liver, early posttransplant, and late follow-up images. (A) Case no. 1 who received a split hyper-reduced left lateral section graft. (B) Case no. 2 who received a left lateral section graft. Multiple hepatic masses were pathologically diagnosed of hemangiomas, but two of them were hepatocellular carcinomas. (C) Case no. 3 who received a split left lateral section graft. (D) Case no. 4 who received a hyper-reduced left lateral section graft. (E) Case no. 5 who received a left lateral section graft. (F) Case no. 6 who received a whole liver graft. The patient died of graft failure and intracerebral bleeding at 35 months after transplantation. (G) Case no. 7 who received a left lateral section graft.
|
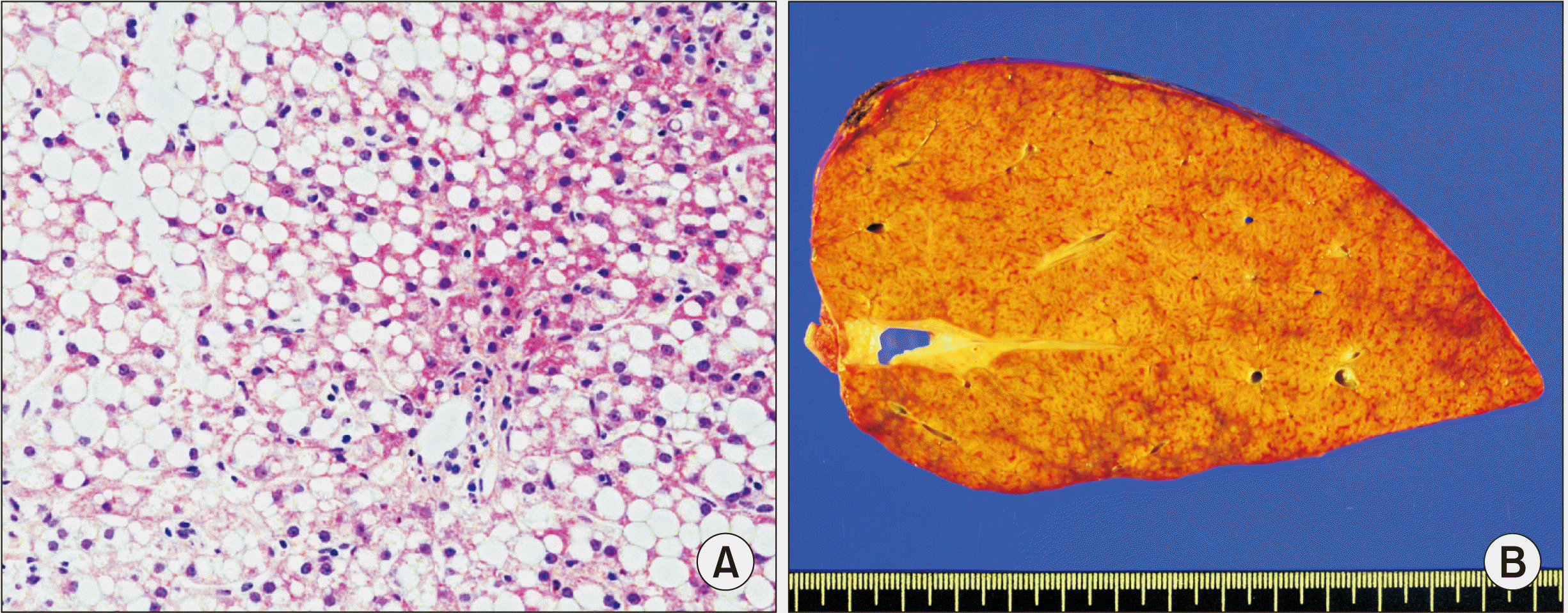
All five patients with PFIC type 2 recovered from the LT operation. They are doing well to date. In contrast, each one patient with PFIC type 1 underwent retransplantation due to graft failure or died due to multi-organ failure. One patient with PFIC type 1 recovered from the LT operation (case no. 3). However, liver enzyme levels were elevated with severe fatty change, mild portal inflammation, severe bile duct damage, and bile ductular proliferation on liver biopsy at posttransplant 4 months (Fig. 2A) [10]. Graft function progressively deteriorated. Retransplantation using a whole liver graft recovered from a 2-year-old deceased donor was performed at 39 months after the first LT (Fig. 2B). The explant liver pathology showed steatohepatitis of stage 3 and grade 3, with severe fatty change (70%–80%), severe bile duct damage, and bile ductular proliferation. The patient is currently doing well with mild fatty change of the liver graft (Fig. 1C) at 47 months after the retransplantation. Another patient with PFIC type 1 recovered from the LT operation (case no. 6), but suffered from cytomegalovirus reactivation, acute cellular rejection, biliary complication, and growth retardation. At 35 months posttransplant, the patient was admitted due to sepsis. During recovery from sepsis, intracerebral hemorrhage occurred and the patient passed away. The primary cause of patient death was graft failure-associated multiorgan failure.
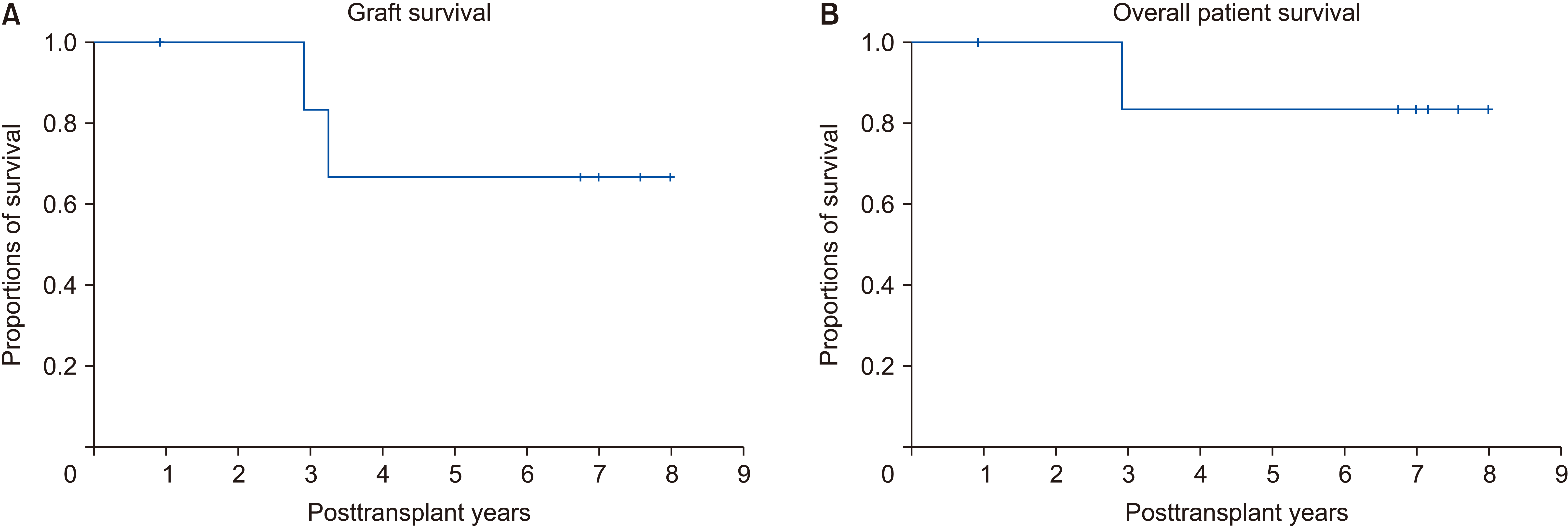
One patient who showed multiple high-enhancement masses in the liver revealed multiple hemangiomas and two small hepatocellular carcinomas (HCCs) (case no. 2). The patient did not show tumor recurrence for 91 months to date. Overall graft and patient survival rates in these seven patients at 5 years were 66.7% and 83.3%, respectively (Fig. 3).
BSEP immunostaining and genetic mutation analysis findings
BSEP immunohistochemical staining in two patients with PFIC type 1 showed normal canalicular expression (Table 2). PFIC type 2 showed focal loss of BSEP expression in two patients and total loss of BSEP expression in three patients (Fig. 4). Results of gene analysis are also presented in Table 2. One patient did not undergo gene study at our institution because PFIC was diagnosed abroad.
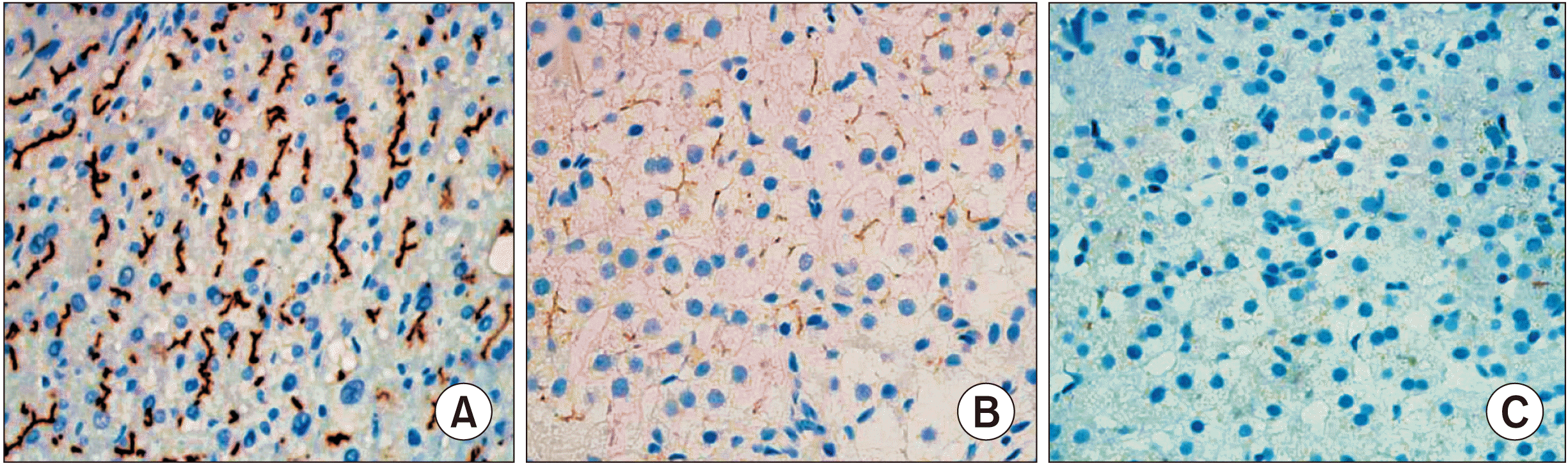 | Fig. 4Immunohistochemical staining for bile salt export pump showing normal canalicular expression (A: normal control patient, ×200), focal loss (B: case no. 4, ×200), and total loss (C: case no. 2, ×200).
|
Table 2
Profiles of bile salt export pump (BSEP) immunostaining and gene analysis
![]()
Go to :
DISCUSSION
LT has been accepted as the only effective treatment for PFIC patients with end-stage liver disease. Indications for LT include severe pruritus, significant growth retardation, liver cirrhosis, and liver failure. In the present study, occurrence of progressive liver failure combined with genetic mutation analysis findings was an indication of LT because the clinical course of PFIC is fatal. PFIC patients usually have growth retardation and metabolic bone disease mainly due to impaired vitamin D and calcium absorption caused by reduced bile secretion. Growth retardation is significantly improved after LT [11,12]. Although PFIC is a well-known disease worldwide, there are only a few case reports and a small case series on PFIC in Korean patients [7,10].
Although identification of a mutation in an associated gene is not mandatory for diagnosis of PFIC, it is an important diagnostic tool [13]. PFIC is a heterogeneous group of liver diseases, although laboratory findings, such as low serum γ-glutamyl transpeptidase despite direct hyperbilirubinemia, are similar in PFIC type 1 and type 2 patients [7]. These findings implicate that hyperbilirubinemia in PFIC is associated with bile excretion by the hepatocytes. On the contrary, common forms of neonatal cholestasis are characterized by increased γ-glutamyl transpeptidase usually due to damaged bile ducts caused by detergent effect of bile salts [14,15].
Approximately 80 and 100 genetic mutations have been identified in PFIC type 1 and type 2 patients, respectively [7]. Although PFICs are autosomal recessive hereditary diseases, compound heterozygous or homozygous mutations might be associated with structural and functional defects. More severe forms of PFIC are likely to be associated with homozygous frame shift and nonsense mutations as well as large genomic deletion. By contrast, benign recurrent intrahepatic cholestasis, a milder form of PFIC, is more likely to be associated with heterozygous missense mutations [16].
The majority of point mutations in PFIC type 1 and type 2 are missense, nonsense, and splicing mutations. Structurally abnormal proteins in PFIC type 1 are due to frame shift, splice site, nonsense mutations, and large genomic deletion. Heterozygous mutations have been associated with good prognosis and low penetrance in PFIC [17]. ABCB11 mutations were not detected in two patients with PFIC type 2 in the present study. Less than 10% of PFIC patients have no or monoallelic mutations. Mutations in these patients might be present in regulatory domains, untranslated regions, and introns that cannot be tested by present assessment methods [18]. Despite the absence of mutations or the presence of a single heterozygous ABCB11 mutation, the diagnostic sensitivity of clinical and pathologic findings with negative BSEP immunostaining is approximately 90% [19]. Therefore, absence of ABCB11 mutations cannot exclude PFIC type 2.
Definitive preoperative diagnosis of PFIC is often difficult if notable gene mutations are not identified. The diagnosis of PFIC type 1 in case no. 6 was based on the pathological and immunohistochemical staining findings as follows: moderate canalicular cholestasis with granular bile; minimal portal inflammation with ductular proliferation; perivenular and periportal fibrosis; loss of CD10 expression; dilated canaliculi filled with coarsely granular bile; and intact BSEP expression. These findings support the diagnosis of PFIC type 1.
Although PFIC is known as an autosomal recessive hereditary disease, four of the seven PFIC cases underwent living donor LT in the present study primarily because of a relatively low incidence of deceased donors in Korea. For allocating split LT, PFIC patients have disadvantages due to low pediatric end-stage liver disease scores in comparison with biliary atresia patients. Some patients had to undergo LT at very young infancy, which made size-matched allocation of a split liver organ more difficult. These were the background reasons why two Korean patients underwent living donor LT. In addition, two of living donor LT cases were international cases, thus they were not allowed for deceased donor LT unless they lived more than one year in Korea.
Ursodeoxycholic acid is considered an initial treatment for patients with PFIC [20]. It can alleviate symptoms in PFIC type 1 patients. However, PFIC type 2 patients generally respond to ursodeoxycholic acid poorly, suggesting that this agent has uncertain effects on the progression of liver disease [21]. LT is the most efficient and the last therapeutic option in patients presenting with liver failure [22,23]. Selection criteria for LT candidates do not differ from those for patients with other liver diseases. Major indications for LT include end-stage liver disease, concurrent HCC, and intractable pruritus with no response to biliary diversion. Although LT can result in remission in 75% to 100% of patients, regardless of PFIC type, specific complications and relapse of disease should be carefully considered prior to LT [11,23,24].
Hori et al. [22,23] studied 14 patients who underwent living donor LT for PFIC (11 cases of PFIC type 1 and 3 cases of PFIC type 2). Three of 11 PFIC type 1 recipients died, while all three PFIC type 2 recipients survived. Eight of these 11 PFIC type 1 recipients developed steatosis after LT. Nine of these 11 PFIC type 1 recipients developed fibrosis after LT. In contrast with PFIC type 1 recipients, PFIC type 2 recipients did not develop steatosis or fibrosis after LT. Liu et al. [12] have reported two cases of PFIC type 1 developing steatohepatitis within 1 year after LT, suggesting the necessity of long-term follow-up. PFIC type 2 patients with some subtypes are prone to recurrence of liver diseases [25]. In the present study, all two patients with PFIC type 1 showed hepatic steatosis. One patient had to undergo retransplantation and the other patient died due to multi-organ failure. In contrast, all five patients with PFIC type 2 showed uneventful posttransplant courses. The surviving retransplant patient with PFIC type 1 has been regularly followed up with ultrasonography due to a high risk of steatohepatitis.
HCC is a very rare disease in infant patients and most HCCs arise in the setting of prior liver disease HCCs [26]. They might be diagnosed incidentally in the explant liver or detected during surveillance of underlying disease. Interestingly, three of four patients who underwent AFP test in the present study showed high elevation of AFP levels before LT. Of them, one patient showed two incidentally detected small HCC masses at the explant liver [27]. We did not take account the possibility of HCC at the time of LT operation because multiple hepatic tumors were considered as infantile hemangioendothelioma. HCC is rarely diagnosed in infant patients with biliary atresia or other disease-associated liver cirrhosis [28-31].
This study has some limitations. First, this study was a small series from a single center. Thus, nationwide or international studies are necessary. Second, gene analysis in this study was not completely conducted. The association between AFP and PFIC needs to be assessed because of the absence of such information in the literature.
In conclusion, LT is currently the only effective treatment for PFIC-associated end-stage liver diseases. However, relapse of disease and other complications including steatohepatitis can occur after LT. Thus, it is mandatory to perform regular posttransplant follow-up, especially for patients with FPIC type 1.
Go to :
 XML Download
XML Download