

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Neuroendocrine tumors (NETs), previously called carcinoid tumors, originate from neuroendocrine cells distributed throughout the body; therefore, they can occur in most organs, but mainly occur in the digestive system, and have a prevalence of approximately 2/100,000.1 Gastrointestinal stromal tumors (GISTs) are mesenchymal neoplasms that can occur in any part of the gastrointestinal tract.2,3 Both NET and GIST are confirmed by biopsy, and their treatment is radical resection.4,5 Since the clinical manifestations of both diseases, such as abdominal pain and intestinal bleeding, may vary, caution should be taken when differentially diagnosing them from other diseases.4,5 We report a case of NET and GIST found metachronously in a patient without symptoms suggestive of a genetic disease.
Go to : 
CASE REPORT
A 41-year-old female with no specific past or family history underwent gastroscopy at a local medical institution because of epigastric pain. Her minor papilla was found to be enlarged, and she was referred to Busan Paik Hospital for further evaluation. At presentation, her vital signs were a blood pressure of 114/69 mmHg, pulse rate of 82 beats per min, respiratory rate of 20 breaths per min and body temperature of 36.5℃. Initial laboratory evaluation revealed a leukocyte count of 4,070/mm3, a hemoglobin level of 11.2 g/dL, and a platelet count of 199,000/mm3. Her CA 19-9 level was found to be less than 0.600 U/mL, and her CEA level was 1.18 ng/mL.. In addition to the minor papilla, the major papilla was found to be enlarged too, and biopsy was performed for each papilla (Fig. 1). Histopathologic examination revealed NET, and PET-CT was performed to check for metastasis. There was no metastasis; therefore, endoscopic papillectomy of the major and minor papillae was performed.6 The patient’s 24-hour urinary 5-hydroxyindoleacetic acid (5-HIAA) excretion was normal (2.2 mg/day); immunochemical staining confirmed a Ki-67 index <2%, indicating grade 1 NET. Endoscopy was performed 6 months after papillectomy, and endoscopy and biliary CT were performed 18 months later, but there were no specific findings. Multiple duodenal and jejunal submucosal nodules were observed on biliary CT and endoscopy performed at the 30 months follow-up, but the patient had no complaints (Fig. 2A-C). Endoscopic ultrasound was performed due to the suspicion of multiple recurrent NETs, and approximately 9×6 mm and 4×3 mm hypoechoic homogenous lesions were seen in the 4th layer (Fig. 2D). Therefore, the patient was referred for surgery. Chromogranin A and 5-HIAA levels were normal (89.1 ng/mL and 2.3 mg/day, respectively). As PET-CT showed no metastasis, pyloruspreserving pancreaticoduodenectomy was performed. Surgical specimen biopsy confirmed the diagnosis of multiple duodenal and jejunal GIST and metastatic NET in the peritumoral lymph node (Fig. 3). Immunohistochemistry showed positive CD34 and C-kit results for the GIST. The metastatic NET was positive for synaptophysin, the Ki-67 index was 1%, and the mitotic count was <1/10 HPF; therefore, it was NET grade 1.
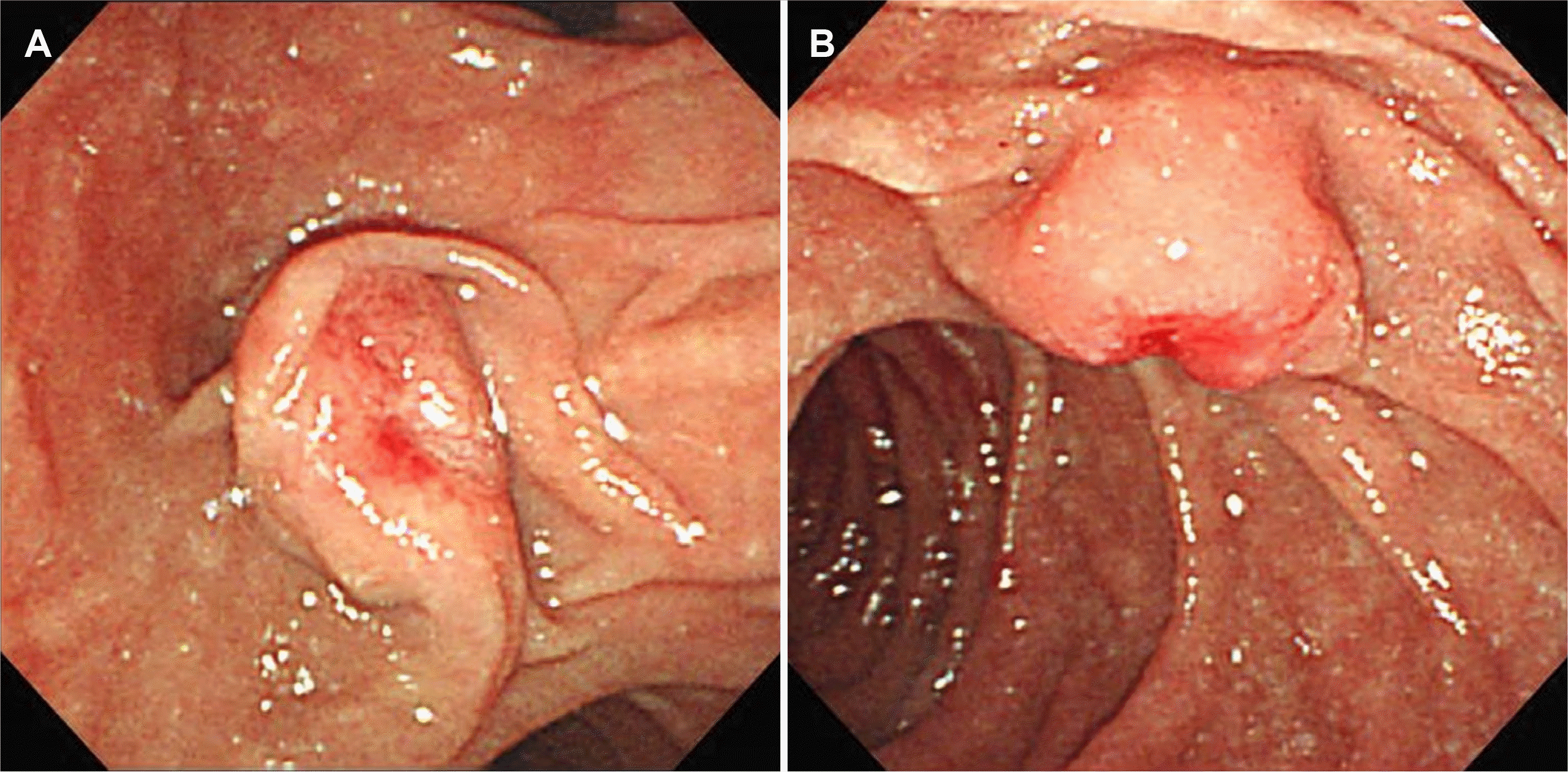 | Fig. 1Endoscopic retrograde choangiopancreatography (ERCP) at the time of initial diagnosis. ERCP reveals (A) major papilla and (B) minor papilla.
|
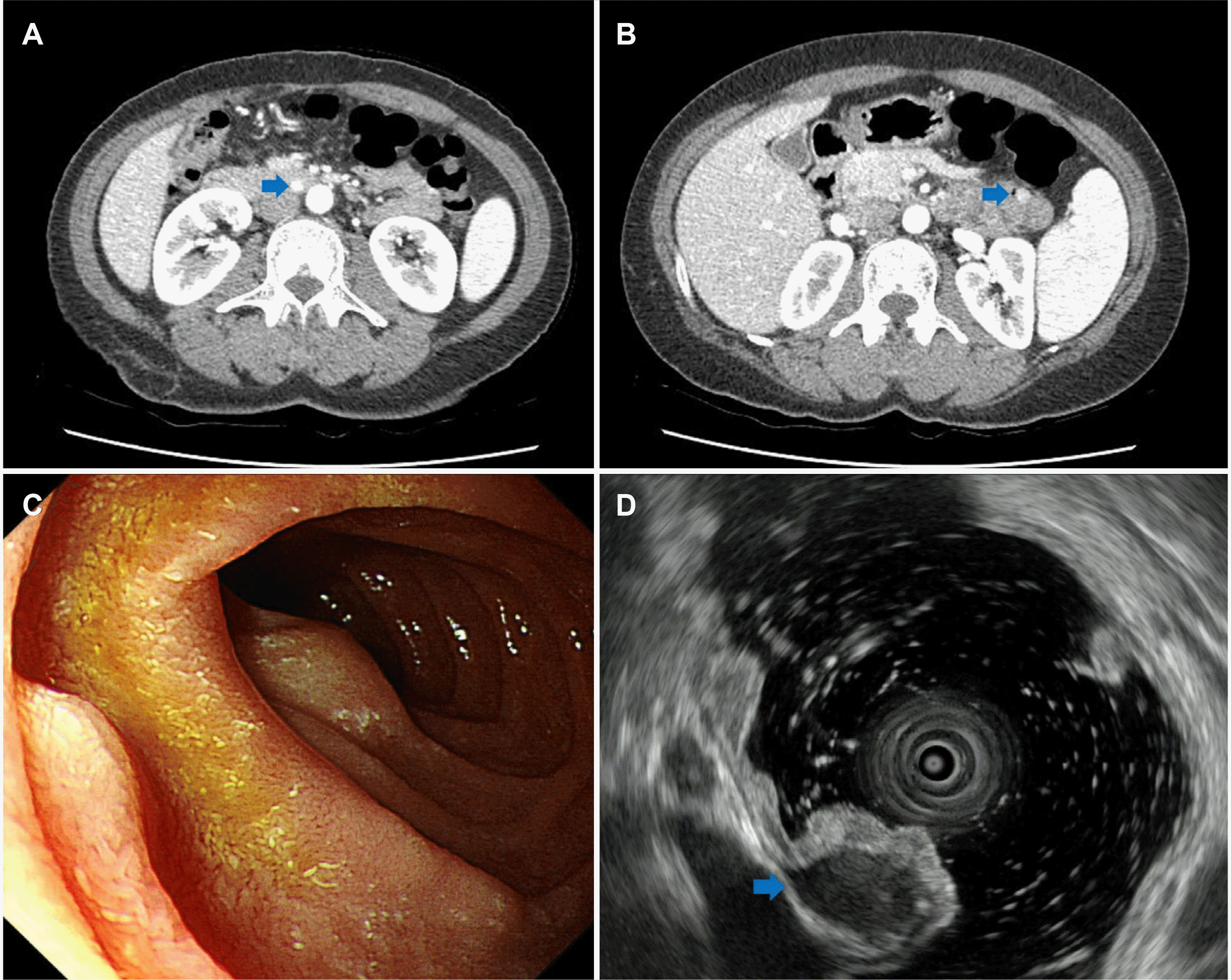 | Fig. 2Biliary computed tomography, endoscopy and endoscopic ultrasound (EUS) findings at follow-up evaluation. (A, B) Submucosal nodules were found in duodenum and jejunum (arrows). (C) Endoscopic image showed polypoid lesions in duodenum. (D) EUS reveals hypoechoic homogenous lesions on the 4th layer (arrow).
|
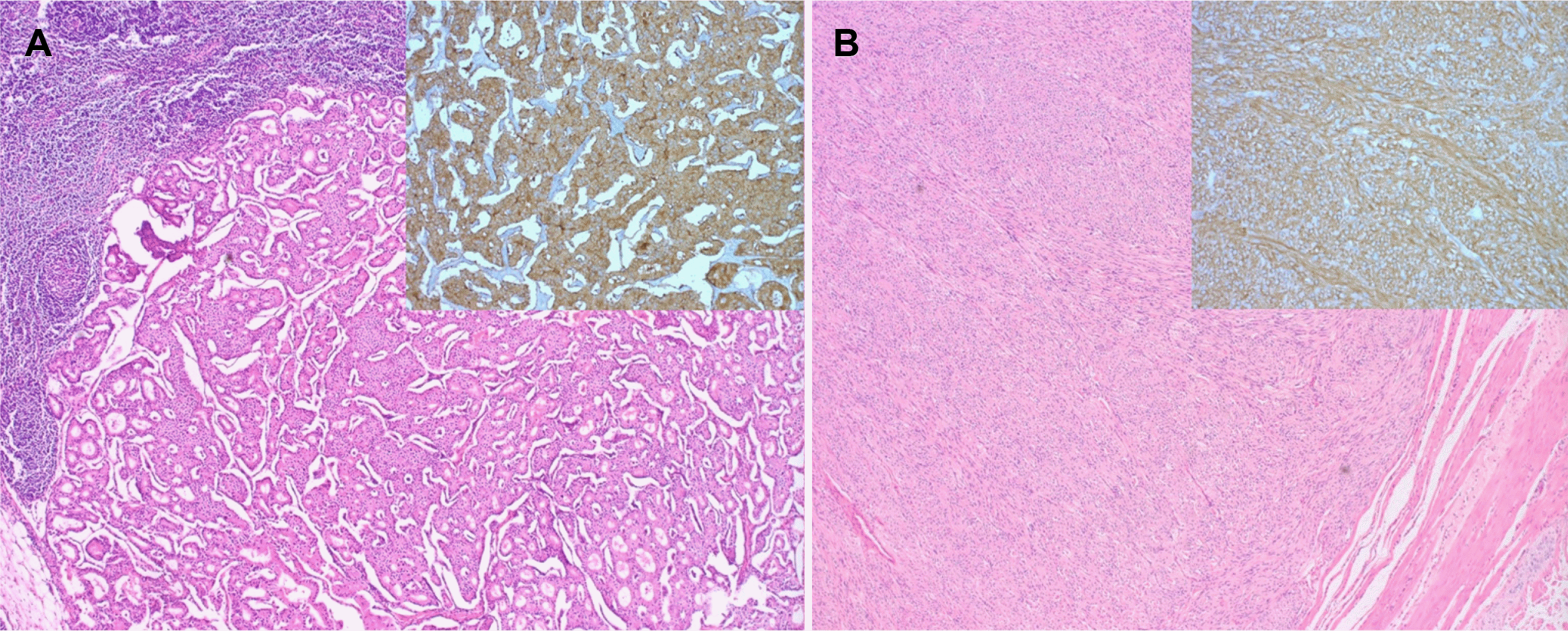 | Fig. 3Histologic features of the (A) neuroendocrine tumor (B) and gastrointestinal stromal tumor. (A) Histopathological evaluation reveals trabecular pattern (hematoxylin and eosin stain [H&E], ×100). Immunohistochemical staining for synaptophysin index is positive (inset). (B) Histopathological evaluation reveals spindle cells proliferation (H&E, ×100). Immunohistochemical staining for C-kit index is positive (inset).
|
Go to :
DISCUSSION
NETs can cause various symptoms, such as hot flashes, diarrhea, intermittent abdominal pain, and gastrointestinal bleeding. According to the presence or absence of symptoms, they are divided into functional and non-functional NETs. Biochemical markers, such as serum chromogranin A (CgA), urinary 5-HIAA, and synaptophysin, are useful in making the diagnosis.7,8 Imaging tests, such as CT, MRI, and PET-CT, are also helpful. Confirmation of the diagnosis is made by biopsy. According to the 2019 WHO standards, NETs are classified into G1-3, small-cell neuroendocrine carcinoma, large-cell neuroendocrine carcinoma, and mixed neuroendocrine-nonneuroendocrine neoplasm by their mitotic rate (mitoses/2 mm2) and Ki-67 index (%).9 Treatment is radical resection, and even if metastasis is present, resection should be considered as much as possible. If surgery is not possible, medications, such as somatostatin analogs, everolimus, and sunitinib, can be used.10-12 NET is usually diagnosed after the disease has already progressed due to its various clinical symptoms. If radical resection is not possible, the prognosis is usually poor.
GIST is thought to arise from the interstitial cells of Cajal that coordinate the movement of the gastrointestinal tract and are located in the muscular lining of the intestinal wall below the epithelium. Mutations in the KIT or PDGFRα genes are known to be important pathological mechanisms, and mutations in the KIT gene are more common, at approximately 80%.13,14 Rarely, GISTs may occur genetically or accompany a specific syndrome, including familial GIST and type 1 neurofibromatosis.15 When it occurs primarily, it occurs most frequently in the stomach (40-60%), followed by the small intestine (30%).16 Metastasis occurs in about 10-20% of patients, and it commonly occurs to the peritoneum and liver.17 GIST may show clinical features, such as gastrointestinal bleeding or abdominal pain, but it is often asymptomatic and discovered incidentally. Although biopsy is essential for diagnosis, it is often not possible to obtain tumor tissue through endoscopy because GIST occurs in the submucosal muscle layer. Therefore, in some cases, tissue is obtained by biopsy simultaneously during treatment by endoscopic ultrasound or laparoscopic surgery. CT is recommended for the initial evaluation of patients with suspected GIST and is an essential tool for staging in patients diagnosed with GIST. In patients for whom it is difficult to perform CT, MRI can be a substitute, and PET-CT can be helpful in staging. The basic treatment is radical resection. Metastatic GIST can be treated with imatinib or sunitinib, but it is difficult to expect a cure, so it is necessary to consider removing all gross lesions through surgery 6 to 20 months after starting drug treatment and to continue imatinib after surgery.18-22
With the exception of some genetic diseases, double primary cancers are rarely diagnosed. In particular, it is even rarer for two tumors with low prevalences to be diagnosed simultaneously. NET and GIST have been reported to be diagnosed simultaneously in the case of neurofibromatosis type 1, but it is known that they are extremely rare in patients with no genetic diseases, as in this case.23,24 Of the previous reports regarding the simultaneous occurrence of GIST and NET in the gastrointestinal tract, we achieved to retrieve data only for four cases. Two cases were diagnosed in patients without complaints and the others were diagnosed in patients with symptoms such as melena or abdominal pain.25-28 NET and GIST often show non-specific symptoms, like the patient in the present case, and since various symptoms appear, it is always difficult to diagnose, unless suspected. As in the previous two reports, it may be diagnosed incidentally without symptoms, so regular health screening is important. If there are suspicious lesions, additional workup or follow-up should be actively performed. In addition, since radical resection is possible only when detected early, NET or GIST should always be considered as a differential diagnosis whenever the patient's symptoms and imaging tests suggest them.
Go to :
 XML Download
XML Download