

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is characterized by chronic airway inflammation as well as progressive and incomplete reversible airflow obstruction [1,2]. Tobacco smoking is an important risk factor for COPD [3-5]. Cigarette smoke extract (CSE) has been shown to inhibit the activity of multidrug resistance protein 1 (MRP1) in human bronchial epithelial (16HBE) cells, slow-down the removal of toxic substances produced by cigarette smoke in cells, and increased the cytotoxicity of CSE [6,7]. Ergosterol, in the CSE-induced 16HBE cells and COPD murine models, can improve the increased CSE-induced inflammatory cytokines and oxidative stress [8]. Stimulation by cigarette smoke leads to oxidative stress in the lungs, which can promote the aggregation of inflammatory cells in the lungs and lead to release of proinflammatory mediators. The latter activate more inflammatory cells to form a “cascade” effect and promote continuous amplification of chronic pulmonary inflammation [9,10]. The chronic inflammatory response and imbalance in oxidative stress influence and promote each other during the occurrence and development of COPD [11]. Therefore, controlling the inflammatory response and regulating the imbalance of oxidative stress are rational approaches in COPD treatment [12].
Glucocorticoids (GCs) are an important treatment modality for COPD because they have powerful anti-inflammatory effects. GCs can recruit histone deacetylase 2 (HDAC2) to gene-expression sites to reduce the degree of histone acetylation and inhibit expression of inflammatory genes. However, some patients are insensitive to GCs, which limits their therapeutic effects [13,14]. The activity and expression of HDAC2 is reduced significantly in COPD patients, which weakens the anti-inflammatory effect of GCs [15-18]. CSE or oxidative stress induced by cigarette smoke induces low corticosteroid reactivity by inhibiting the activity and expression of HDAC2 [19,20]. Curcumin reverses the sterol insensitivity induced by CSE or oxidative stress by maintaining the activity and expression of HDAC2 [21].
4-hydroxynonenal (4-HNE) is the most common toxic product of lipid peroxidation during oxidative stress. It can bind covalently with HDAC2 to destroy protein function, thereby leading to HDAC2 inactivation [22-25]. Studies have shown that the decreased activity and expression of HDAC2 lead to the up-regulated expression of nuclear factor-kappa B (NF-kB) and the increased release of tumor necrosis factor (TNF)-α and interleukin (IL)-8 [26,27]. In addition, the increased expression of HDAC2 can inhibit the expression of inflammatory genes through the c-Jun/PAI-1 pathway, and reduce the inflammatory response induced by lipopolysaccharide (LPS), thus playing an important part in immune defense [28]. Sulforaphane is an agonist of nuclear factor E2-related factor 2 (Nrf2). Sulforaphane, as an agonist of Nrf2, which can denitrogenate HDAC2, restored the sensitivity of alveolar macrophages to dexamethasone (DEX), and reversed the GC resistance in COPD patients [29]. Therefore, exploration of pathways to reverse GC resistance and enhance the anti-inflammatory effect of GCs in COPD is important.
The Nrf2-antioxidant responsive element (ARE) pathway is one way the body can inhibit the effects of exogenous stimuli and oxidative damage [30], which is associated with COPD development [31]. Nrf2 activation and the upregulation of its target gene not only counteract oxidative stress, but also limit the severity of inflammatory damage [32]. Binding of Nrf2 to ARE in the nucleus initiates the expression of a series of phase-II detoxification enzymes, antioxidant genes, and export proteins [33-35], such as heme oxygenase-1 (HO-1), glutathione transferase, and MRP1. As one of the downstream target molecules of Nrf2, HO-1 reduces the overall formation of reactive oxygen species (ROS) by degrading heme, producing carbon monoxide and biliverdin/bilirubin, and releasing free iron. Therefore, the Nrf2/HO-1 signaling pathway is an important antioxidant defense system against various diseases [36]. Brusatol is a specific inhibitor of Nrf2. Brusatol can block Nrf2 activity, reduce the upregulation of HO-1 expression mediated by punicalagin, and weaken the antioxidant and anti-inflammatory effects of punicalagin [37]. MRP1 shows high expression in lung tissues and plays an important part in protecting tissues from toxicity [38,39]. In addition to mediating adenosine triphosphate-dependent extravasation of endogenous and exogenous drugs, the protective mechanism of MRP1 also mediates the extravasation of sulfate, glucuronic acid, and glutathione (GSH) conjugates, including complexes formed by binding 4-HNE to GSH [40], which may reduce the damage of 4-HNE to HDAC2. Also, MRP1 can mediate efflux of leukotriene-C4 and improve allergic airway inflammation [41]. Studies have shown that allyl isothiocyanate can improve the lung function of COPD-model rats by upregulating MRP1 expression via the Nrf2 signaling pathway, which has a therapeutic effect upon COPD [42,43].
Metformin (MET) (Fig. 1) is one of the most widely used oral hypoglycemic drugs worldwide. It is front-line treatment of type-2 diabetes mellitus. Recently, it has been shown that MET not only improves insulin resistance [44] but also has anti-cancer [45], anti-inflammatory [46,47] and other activities. Studies have revealed MET to have an anti-inflammatory role because it reduces the level of proinflammatory factors, increases the level of anti-inflammatory factors, and inhibits oxidative stress [48]. Thus, MET may have a certain therapeutic effect upon various inflammatory diseases. Another study showed that MET could reduce the release of mitochondrial ROS and IL-6 in alveolar macrophages exposed to particulate matter of diameter ≤ 2.5 µm (PM2.5) [49]. The latest research suggests that MET and silymarin have a protective effect upon cyclosporine A-induced hepatorenal toxicity because they restore oxidative homeostasis and regulate the release of proinflammatory mediators [50]. Those findings provide a theoretical basis for MET in COPD treatment. Therefore, whether MET can improve GC resistance in COPD conditions merits further study.
Through establishment of a COPD model and a model of GC resistance, combined with in vivo and in vitro experiments, we explored if MET can activate the Nrf2/HO-1 signaling pathway, enhance the function and expression of MRP1, and then promote HDAC2 expression. We sought to provide a new strategy for COPD treatment and alleviation of GC resistance.
Go to : 
METHODS
Drugs and reagents
MET was purchased from Fengtai Weiyuan Technology Co., Ltd. (Wuhan, China). N-Acetylcysteine (NAC) and DEX were obtained from Sigma-Aldrich (Saint Louis, MO, USA). LPS and Cell Counting Kit-8 (CCK-8) kits were supplied by Solarbio (Beijing, China). Cigarettes were provided by Anhui China Tobacco Industry Co., Ltd. (Anhui, China). Assay kits for bicinchoninic acid (BCA) protein, total superoxide dismutase (SOD), lipid-peroxidation malondialdehyde (MDA), glutathione peroxidase (GSH-Px), and the MRP1 inhibitor MK571 were obtained from Beyotime Institute of Biotechnology (Shanghai, China). The HO-1 inhibitor tin protoporphyrin (SnPP) was obtained from MedChemExpress (Monmouth Junction, NJ, USA). Enzyme-linked immunosorbent assay (ELISA) kits for IL-8 (rat), TNF-α (rat), and IL-8 (human) were supplied by Meimian Industrial (Jiangsu, China). Antibodies against MRP1, Nrf2, and HO-1 were purchased from Abcam (Cambridge, UK). Anti-β-actin and horseradish peroxidase-conjugated secondary antibodies were obtained from Zhongshan Jinqiao Biotechnology (Beijing, China).
Animals and grouping
The protocol for animal experiments was approved by the Experimental Animal Ethics Committee of Anhui University of Chinese Medicine (Hefei, China) and conformed to ethical standards accepted internationally. All rats received humane care. Male specific pathogen-free rats of the Sprague–Dawley strain (180–220 g) were housed at room temperature (25 ± 1°C) under a 12-h light–dark cycle , and had free access to food and water.
Fifty healthy Sprague–Dawley rats were divided randomly into five groups of 10: A (control), B (COPD model), C (MET low dose), D (MET high dose), and E (NAC). LPS combined with fumigation was used in all groups except the control group. On days 1, 15, 30, and 45 day of modeling, LPS solution was injected into the airways of rats. On the other days, each rat was placed in an inhalation chamber, allowed to smoke one cigarette for 1 h at a time, twice a day, at an interval of > 6 h. After cessation of smoking, the rat began to undergo gavage on day-61 for 15 consecutive days. Rats in groups C and D were given MET at 150 and 300 mg/kg/d, respectively. Rats in group E were administered NAC (800 mg/kg/d). Rats in groups A and B underwent instillation with an equal amount of solvent (vehicle).
Dose determination of MET and NAC was based on a study on the toxicity and toxicokinetics of MET in rats. Rats were given MET at different doses for 13 weeks. It was found that administration of ≥ 900 mg/kg/d resulted in death. Weight loss and mild metabolic acidosis were associated with administration of ≥ 600 mg/kg/d [51]. In a study on whether MET can maintain mucosal integrity in an experimental model of colitis by inhibiting oxidative stress and proinflammatory signaling, MET was administered at a low dose (50 mg/kg) and high dose (500 mg/kg) [52]. By referring to the results stated above, we modified our study design so that 150 mg/kg/d was used for the low-dose group and 300 mg/kg/d for the high-dose group.
The antioxidant capacity of NAC plays an important part in COPD treatment [53]. Studies have shown that a high concentration of NAC (973 mg/kg/d) for 28 days can prevent CSE-induced epithelial thickening and an increase in the number of secretory cells. Low-dose NAC has weak anti-inflammatory and antioxidant effects in COPD treatment [54]. It has been shown that NAC (800 mg/kg/d) can partly reverse the decrease in secretion of vascular endothelial growth factor and expression of vascular endothelial growth factor receptor-2 protein in COPD rats as well as alleviate lung injury, emphysema, and apoptosis of alveolar septal cells [55]. Referring to NAC dose in the literature and our previous laboratory studies, we ensured that the NAC dose in the present study was 800 mg/kg/d [56].
Cells and cell culture
Human bronchial epithelial cells 16HBE were obtained from Shanghai Sixin Biological Technology (Shanghai, China). They were maintained in high-glucose Dulbecco’s modified Eagle’s medium (HyClone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) and 1% penicillin–streptomycin (Beyotime Institute of Biotechnology), and cultured in a humidified incubator at 37°C in an atmosphere of 5% CO2. Cells in each group were treated with different drugs for 2 h after CSE stimulation for 24 h. Then, they were given DEX (10−10 to 10−6 mol/L) for incubation for 1 h and LPS (1 mg/L) for 24 h.
Cytotoxicity and viability assays
We used CCK-8 to calculate the percentage of surviving cells. We inoculated cells in 96-well plates at 5 × 104 cells/ml, followed by treatment with MET (0.0, 2.5, 5, 10, 20, 40, 80 mM) for 12, 24, or 48 h. Afterwards, CCK-8 solution (10 µl) was added to the newly changed medium for an additional 1 h. Finally, the cell viability of the solution in each well was measured with a microplate reader (Multiskan GO; Thermo Scientific, Waltham, MA, USA) at 450 nm.
Determination of lung function
After 15 days of intragastric administration, rats in each group were anesthetized. Then, tracheotomy and endotracheal intubation were undertaken using AniRes 2005 (BestLab, Beijing, China) to detect lung function. Parameters were recorded by software automatically, including forced expiratory volume at 0.3 seconds/forced vital capacity (FEV0.3/FVC%), maximum intermediate expiratory flow (FEF25–75), maximal mid-expiratory flow (MMF), and peak expiratory flow (PEF).
Pulmonary histopathology
Left-lung tissues were immobilized in 4% paraformaldehyde for 24 h, embedded in paraffin, and made into paraffin sections of thickness 4 µm. The paraffin sections were stained by hematoxylin–eosin (H&E) and sealed with neutral gum. Finally, paraffin sections were viewed at 200× magnification under a light microscope. The average score of airway inflammation and Alveolar Damage Index were used to evaluate the degree of infiltration of inflammatory cells in lung tissueto. The scope and severity of lung damage was graded on a scale from 0.5 to 4 (0.5 = “minor”; 1 = “mild”; 2 = “moderate”; 3 = “severe”; 4 = “very severe”) [57].
Immunohistochemistry test
Paraffin sections were blocked with goat serum and then incubated with the primary antibodies against Nrf2, HO-1, and MRP1 overnight at 4°C, respectively. Sections were incubated with peroxidase-conjugated secondary antibodies for 30 min at 37°C. Subsequently, the sections were stained with diaminobenzidine and counterstained by hematoxylin. Eventually, images were captured by a microscope at 400× magnification.
Real-time reverse transcription-quantitative polymerase chain reaction
Real-time reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was applied to measure mRNA expression of HDAC2. Total cellular RNA was isolated from human bronchial epithelial cells 16HBE cells using TRIzol Reagent according to manufacturer (Invitrogen, Carlsbad, CA, USA) instructions. The RNA content of samples was quantified by measuring the absorbance at 260 nm. Complementary (c)DNA was generated using the High-Capacity cDNA Archive kit (Thermo Fisher Scientific) according to manufacturer instructions. Real-time PCR was done with the SYBR Green Master Mix system (Applied Biosystems, Foster City, CA, USA) following manufacturer instructions using an ABI 7500 instrument (Applied Biosystems). The thermocycling conditions comprised pre-denaturation at 94°C for 30 sec, followed by 40 cycles of 94°C for 5 sec (denaturation), and 60°C for 35 sec (annealing and extension). Gene expression was quantified using β-actin as an internal standard, and we calculated the relative expression of mRNA using the comparative 2–ΔΔCT method. RT-qPCRs were done in duplicate. The primer sequences from humans used (forward and reverse, respectively) were 5´-GTCTGCTACTACTACGACGG-3´ and 5´-TTGTCATTTCTTCGGCAGTG-3´ for HDAC2, and 5´-CAAGCAGGAGTACGATGAGT-3´ and 5´-AAAACGCAGCTCAGTAACAG-3´ for β-actin.
Western blotting
A mixture of RIPA buffer (Beyotime Institute of Biotechnology) with 1% phenylmethylsulfonyl fluoride was added into right-lung tissue or 16HBE cells to extract the total protein. The concentration of total protein was measured using a BCA kit. Samples of total protein from each group were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (Sigma-Aldrich) and transferred onto polyvinylidene difluoride (PVDF) membranes. The latter were sealed with 5% skimmed milk for 2 h at room temperature and then, respectively, incubated with primary antibodies overnight at 4°C: anti-Nrf2 (1:1,000 dilution), anti-HO-1 (1:2,000), anti-MRP1 (1:50), anti-HDAC2 (1:2,000), or anti-β-actin (1:2,000). The next day, PVDF membranes were rinsed with phosphate-buffered saline and incubated with peroxidase-conjugated secondary antibodies (1:5,000) for 2 h at room temperature on a shaker. Finally, the PVDF membranes were coated with electrochemiluminescence luminescence solution (Solarbio) evenly in a dark room and exposed in a gel imager. β-actin was used as the quantitative internal reference. Gray values for proteins were analyzed with Image J (National Institutes of Health, Bethesda, MD, USA).
Determination of inflammatory factors and indexes of oxidative stress
The levels of IL-8 and TNF-α, the activity of GSH-Px and SOD, and MDA content in right-lung tissue were determined using the respective ELISA kits according to manufacturer protocols. The absorbance values were measured at the corresponding wavelengths using a microplate analyzer.
In in vitro cell experiments, a semi-logarithmic curve was drawn according to the percent inhibition of IL-8 corresponding to different concentrations of DEX in each group, and the half-maximal inhibition concentration of DEX (IC50-DEX) was calculated using SPSS 24.0 (IBM, Armonk, NY, USA). The effect of MET, SnPP (5 µg/ml) [58] and MK571 (20 µM) [7] on GC resistance induced by CSE was demonstrated by comparing the IC50-DEX values of each group.
Statistical analyses
Data are the mean ± standard deviation. All samples were tested to ascertain if they had a normal distribution. The Student’s t-test was used for comparisons of two independent groups. One-way ANOVA was used for data comparison among multiple groups, and the Student–Newman–Keuls q-test was used for further pairwise comparison. Statistical analyses were undertaken with SPSS 24.0 and p < 0.05 was deemed significant.
Go to :
RESULTS
MET improves the lung function and pathological injury in COPD
Lung function is an important evaluation criterion for establishment of a COPD model. Compared with the control group, exposure to LPS and CSE reduced FEV0.3/FVC%, FEF25–75, MMF, and PEF obviously in the model group, observations which are consistent with the pathological characteristics of chronic bronchitis and obstructive emphysema (Table 1). After treatment with MET (150, 300 mg/kg) and NAC (800 mg/kg), these indicators increased compared with those in the model group. Furthermore, MET improved the indices of lung function in a dose-dependent manner. The results of H&E staining are shown in Fig. 2. In the control group of rats, the structure of bronchioles and alveoli was complete, and few inflammatory cells were observed. In the model group, the bronchial structure was incomplete, alveoli were damaged to varying degrees, and the surface epithelium of the lung was infiltrated by inflammatory cells, all of which are in accordance with the characteristic pathological changes seen in COPD. Treatment with MET offered a remarkable improvement, with a complete alveolar structure and fewer inflammatory cells compared with that in the model group. Taken together, these observations indicated that we had established a COPD model, and that MET could protect against CS- and LPS-induced pathological injury.
 | Fig. 2Histopathological analysis of the lung in rats.(A) Pathological changes in the lung tissues (Scale bars: 100 μm). The magnification is indicated with arrows. (B) Average score of airway inflammation. (C) Alveolar damage index (DI). Data are expressed as means ± SD. MET, metformin; NAC, N-Acetylcysteine. *p < 0.01 vs. control group; #p < 0.05 or ##p < 0.01 vs. model group (n = 3).
|
Table 1
Changes in the lung function in all groups (n = 6)
| Group | FEV0.3/FVC% | FEF25–75 | MMF | PEF |
|---|---|---|---|---|
| Control | 90.278 ± 3.275 | 38.927 ± 4.728 | 44.621 ± 3.098 | 40.449 ± 3.511 |
| Model | 44.039 ± 4.871* | 18.475 ± 2.931* | 20.934 ± 2.550* | 20.502 ± 1.868* |
| MET-150 | 60.685 ± 5.533# | 25.470 ± 2.423# | 29.548 ± 2.826# | 26.159 ± 2.496# |
| MET-300 | 76.089 ± 3.964## | 31.487 ± 3.018## | 35.803 ± 2.461## | 32.478 ± 2.885## |
| NAC | 86.048 ± 7.197## | 36.627 ± 2.594## | 42.887 ± 1.752## | 38.655 ± 3.164## |
Values are presented as mean ± SD. FEV0.3/FVC%, forced expiratory volume at 0.3 seconds/forced vital capacity; FEF25–75, maximum intermediate expiratory flow; MMF, maximal mid-expiratory flow; PEF, peak expiratory flow; MET, metformin; NAC, N-Acetylcysteine. *p < 0.01 vs. control group; #p < 0.05 or ##p < 0. 01 vs. model group.
![]()
MET reduces the inflammatory and oxidative stress in COPD
Expression of IL-8 and TNF-α in rat lung tissue was measured to investigate if MET regulated the inflammatory process in COPD. Inflammatory cells release various cytokines, including IL-8 and TNF-α, which play an important part in the inflammatory response. IL-8 (Fig. 3A) and TNF-α (Fig. 3B) levels increased sharply after exposure to LPS and CS compared with those in the control group. Compared with the model group, IL-8 and TNF-α levels fell markedly after MET treatment. We showed above that MET could inhibit expression of CS- and LPS-induced inflammatory factors. To determine the effect of MET on changes in oxidant–antioxidant systems, the activity of GSH-Px (Fig. 3C) and SOD (Fig. 3D) and MDA content (Fig. 3E) in rat lung tissue were measured. The activity of GSH-Px and SOD in the model group was distinctly lower than that in the control group. However, the activity of GSH-Px and SOD was increased significantly after MET treatment compared with that in the model group. Contrary to the antioxidant enzymes GSH-Px and SOD, MDA is a major marker of oxidative damage. In comparison with the control group, the MDA level in the model group was increased markedly. MDA content decreased upon treatment with MET and NAC compared with that in the model group. These results indicated that MET may attenuate oxidative stress in COPD.
 | Fig. 3Effects of MET on inflammatory factors and oxidative stress parameters in lung tissue of COPD rats.(A) Effect of MET on the level of IL-8 in lung tissues of COPD rats. (B) Effect of MET on the level of TNF-α in lung tissues of COPD rats. (C) Effect of MET on the activity of GSH-Px in lung tissues of COPD rats. (D) Effect of MET on the activity of SOD in lung tissues of COPD rats. (E) Effect of MET on the level of MDA in lung tissues of COPD rats. Data are expressed as means ± SD. MET, metformin; NAC, N-Acetylcysteine; COPD, chronic obstructive pulmonary disease; IL, interleukin; GSH-Px, glutathione peroxidase; SOD, superoxide dismutase; MDA, malondialdehyde; TNF, tumor necrosis factor. *p < 0.01 vs. control group; #p < 0.05 or ##p < 0.01 vs. model group (n = 5).
|
MET upregulates the expression of MRP1 and HDAC2 by activating Nrf2/HO-1 axis
We wished to assess if MET enhanced MRP1 expression through the Nrf2 signaling pathway. Hence, we measured protein expression of Nrf2, HO-1, and MRP1 in lung tissues by immunohistochemical staining and western blotting. Nrf2, HO-1, and MRP1 were expressed mainly in the lung bronchus and alveolar space of rats (Fig. 4A). Software designed specifically for immunohistochemical analyses was used to calculate the integral optical density: protein expression of Nrf2, HO-1, and MRP1 in the model group was reduced dramatically compared with that in the control group. MET treatment increased the protein expression of Nrf2, HO-1, and MRP1 (Fig. 4B). Consistent with the results of immunohistochemical analyses, western blotting (Fig. 5) showed that MET increased the protein expression of Nrf2, HO-1, MRP1, and HDAC2 compared with that in the model group, whereas expression of these proteins was reduced significantly in the model group compared with that in the control group. These observations demonstrated that expression of Nrf2 and its downstream protein HO-1 was positively correlated with MRP1 expression. Therefore, we speculated that MET may increase MRP1 expression by activating the Nrf2/HO-1 signaling pathway. Moreover, expression of HDAC2 protein in lung tissues of rats was consistent with that of Nrf2, HO-1, and MRP1, which suggested that MET could also induce HDAC2 expression in COPD.
 | Fig. 4Immunohistochemical analysis of lung tissues in rats.(A) The immunohistochemistry results of MRP1, Nrf2, and HO-1 proteins in each group (scale bars: 50 μm). The magnification is indicated with arrows. (B) The IOD of MRP1, Nrf2, and HO-1 proteins in each group were analyzed. Data are expressed as means ± SD. Nrf2, nuclear factor E2-related factor 2; HO-1, heme oxygenase-1; MRP1, multidrug resistance protein 1; IOD, integral optical density; MET, metformin; NAC, N-Acetylcysteine. *p < 0.01 vs. control group; #p < 0.01 vs. model group (n = 5).
|
 | Fig. 5Effects of MET on protein expression of Nrf2, HO-1, MRP1, and HDAC2 in lung tissues of COPD rats.Data are expressed as means ± SD. Nrf2, nuclear factor E2-related factor 2; HO-1, heme oxygenase-1; MRP1, multidrug resistance protein 1; MET, metformin; NAC, N-Acetylcysteine; HDAC2, histone deacetylase 2; COPD, chronic obstructive pulmonary disease. *p < 0.01 vs. control group; #p < 0.01 vs. model group (n = 3).
|
MET alleviates the GC resistance in 16HBE cells
A MET concentration of 20 mM and incubation duration of 24 h were selected as conditions in subsequent experiments on cell proliferation using CCK-8 (Fig. 6). IC50-DEX was used as an index of corticosteroid sensitivity, and was determined based on the inhibition of IL-8 release at different DEX concentrations (10−10 to 10−6 mol/L). IL-8 secretion was measured by ELISAs. IC50-DEX of the CSE group was increased dramatically compared with that in the control group. IC50-DEX was reduced in groups pretreated with MET and DEX compared with that in the CSE group. A combination of MET and DEX decreased IC50-DEX significantly compared with that using MET alone or DEX alone. These results indicated that 16HBE cells treated with CSE were less sensitive to DEX compared with 16HBE cells in the control group, which demonstrated that CSE stimulated 16HBE cells to develop GC resistance. However, sensitivity was increased in cells pretreated with MET alone or DEX alone. Furthermore, combination therapy showed an additive effect to alleviate GC resistance (Fig. 7, Table 2).
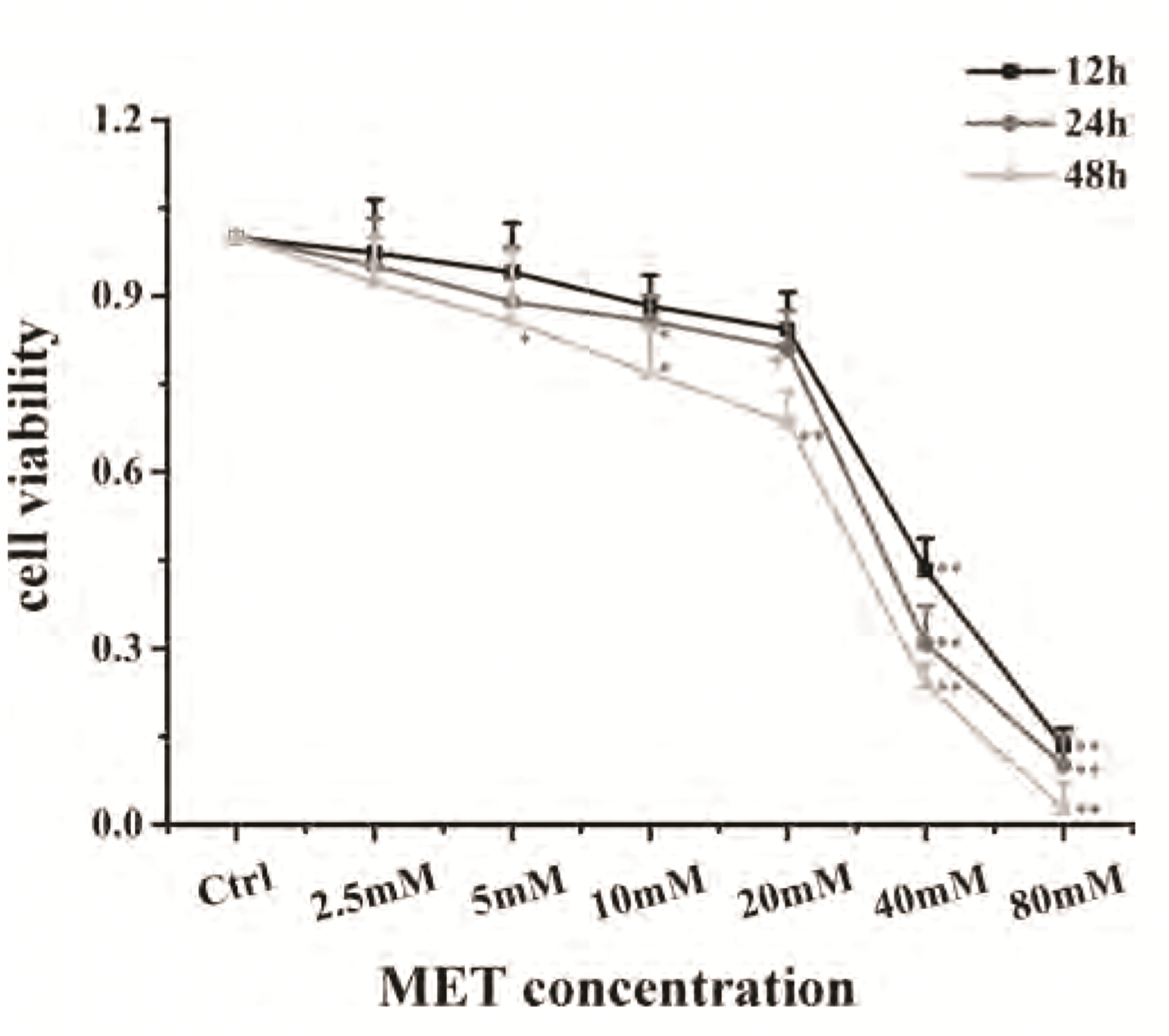 | Fig. 6Survival of 16HBE in the presence of different concentrations of MET for 12, 24, and 48 h.Data are expressed as means ± SD. MET, metformin; 16HBE, human bronchial epithelial. *p < 0.05 or **p < 0.01 vs. control group (n = 3).
|
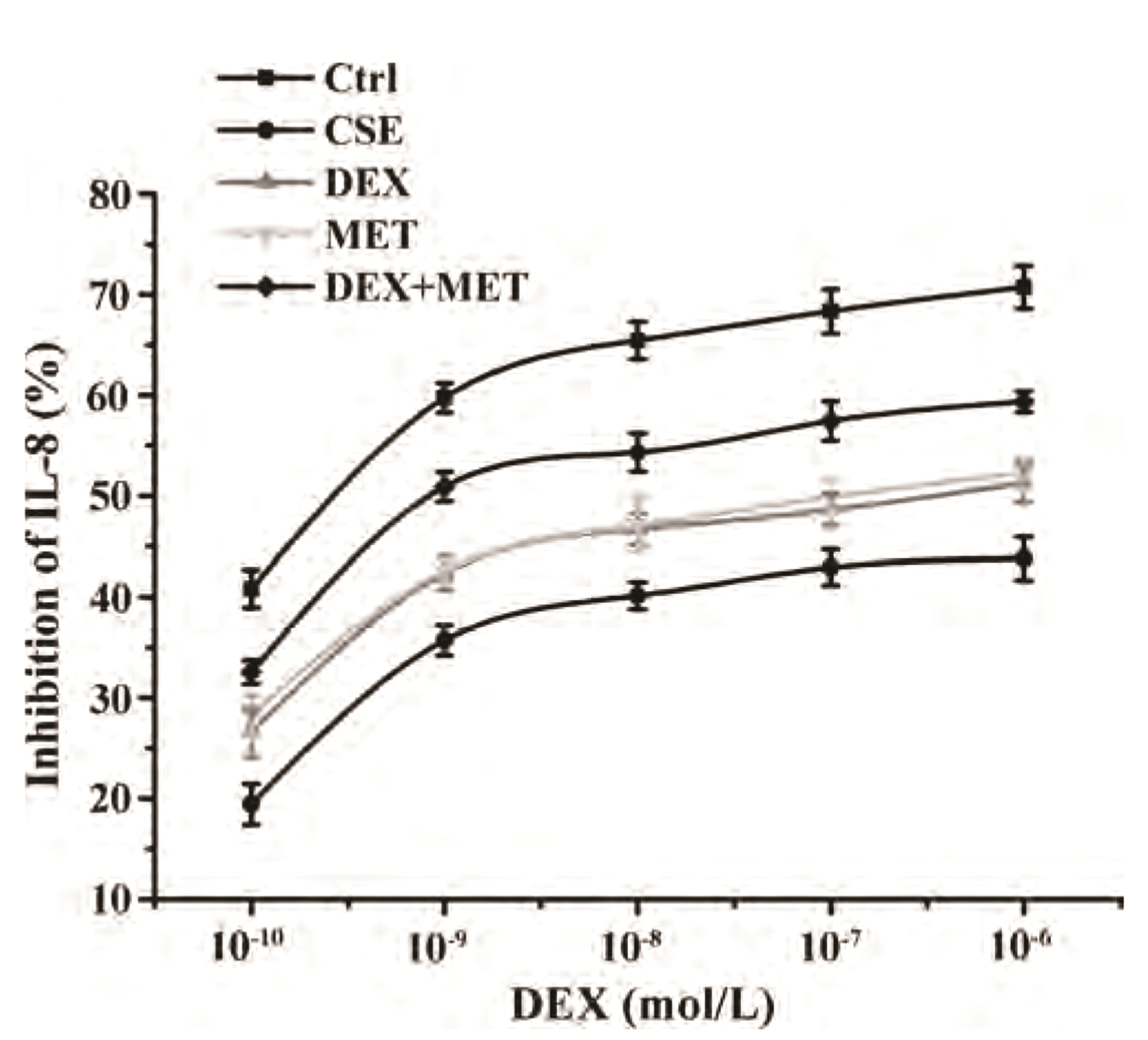 | Fig. 7Effect of MET on IL-8 inhibition rates in each group.Data are expressed as means ± SD (n = 3). MET, metformin; IL, interleukin; CSE, cigarette smoke extract; DEX, dexamethasone.
|
MET enhances the expression of MRP1 and HDAC2 proteins in GC-resistant 16HBE cells via the Nrf2/HO-1 axis
The results shown above suggested that MET had an important role in reducing GC resistance. To ascertain its basic mechanism of action, the protein expression of Nrf2, HO-1, MRP1, and HDAC2 in the cells of each group was measured by Western blotting. Protein expression of Nrf2, HO-1, MRP1, and HDAC2 decreased significantly in the cells of the CSE group compared with those of the control group (Fig. 8). In addition, protein expression of Nrf2, HO-1, MRP1, and HDAC2 in the DEX group, MET group, and combined-treatment group increased notably in contrast with that in the CSE group, whereas protein expression of Nrf2, HO-1, MRP1, and HDAC2 in the combined group was increased obviously compared with that in the DEX group and MET group. Our data demonstrated that the effect of MET on reducing GC resistance may be related to activation of the Nrf2/HO-1 signaling pathway to upregulate MRP1 expression, followed by further regulation of expression of HDAC2 protein.
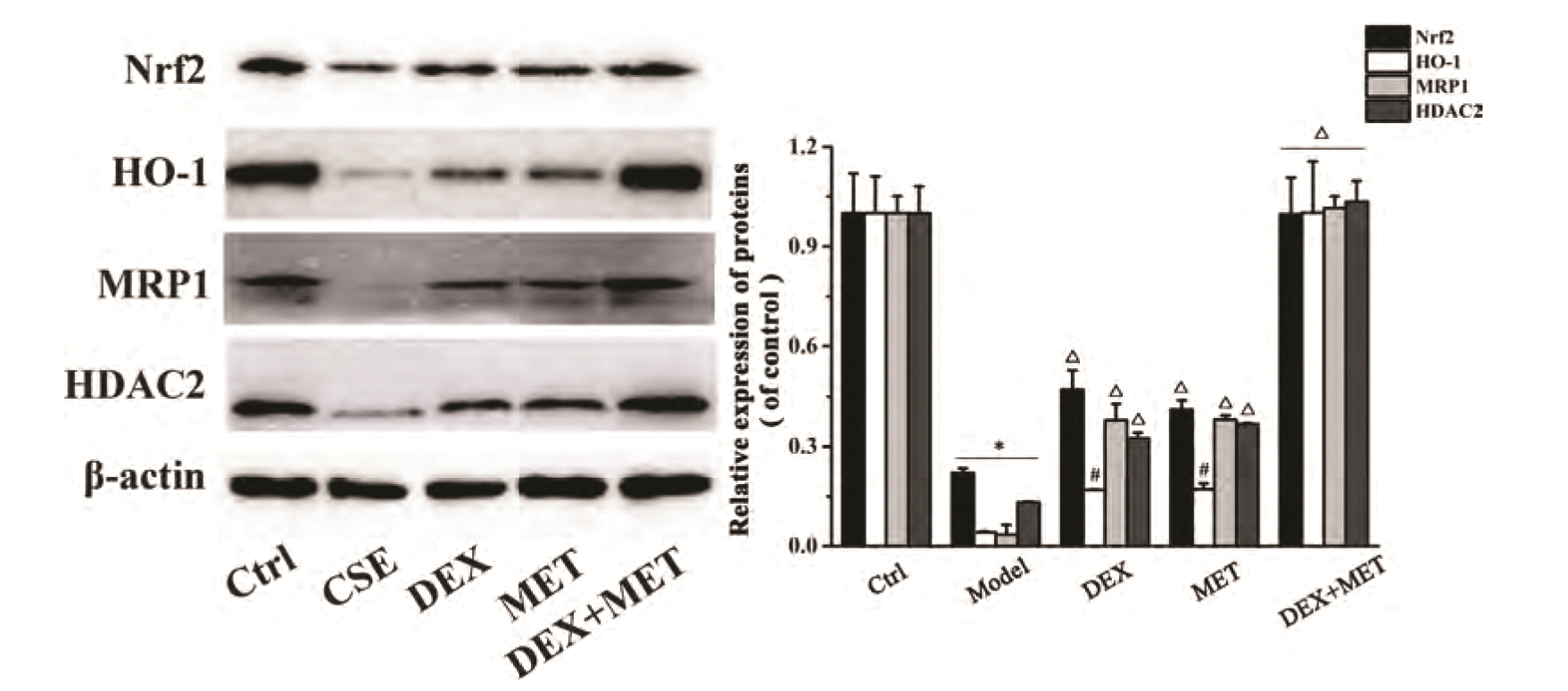 | Fig. 8Effects of MET on the expression of Nrf2, HO-1, MRP1, HDAC2 proteins in cells with glucocorticoid resistance.Data are expressed as means ± SD. Nrf2, nuclear factor E2-related factor 2; HO-1, heme oxygenase-1; MRP1, multidrug resistance protein 1; MET, metformin; HDAC2, histone deacetylase 2; CSE, cigarette smoke extract; DEX, dexamethasone. *p < 0.01 vs. control group; #p < 0.05 or Δp < 0.01 vs. CSE group; Δp < 0.01 vs. DEX group or MET group (n = 3).
|
SnPP weakens the effect of MET to reduce GC resistance
We wished to explore the role of HO-1 in the reduction of GC resistance by MET in 16HBE cells. The latter were incubated with the HO-1 inhibitor SnPP, and the IL-8 concentration in supernatants was measured by ELISAs to obtain IC50-DEX. Compared with the control group, IC50-DEX in the CSE group was increased significantly. IC50-DEX of the combined-treatment group was significantly lower than that of the CSE group. Compared with the combined-treatment group, SnPP-pretreated cells could inhibit the decrease of IC50-DEX significantly. In conclusion, the HO-1 inhibitor SnPP attenuated the ability of MET to reverse GC resistance (Fig. 9, Table 3).
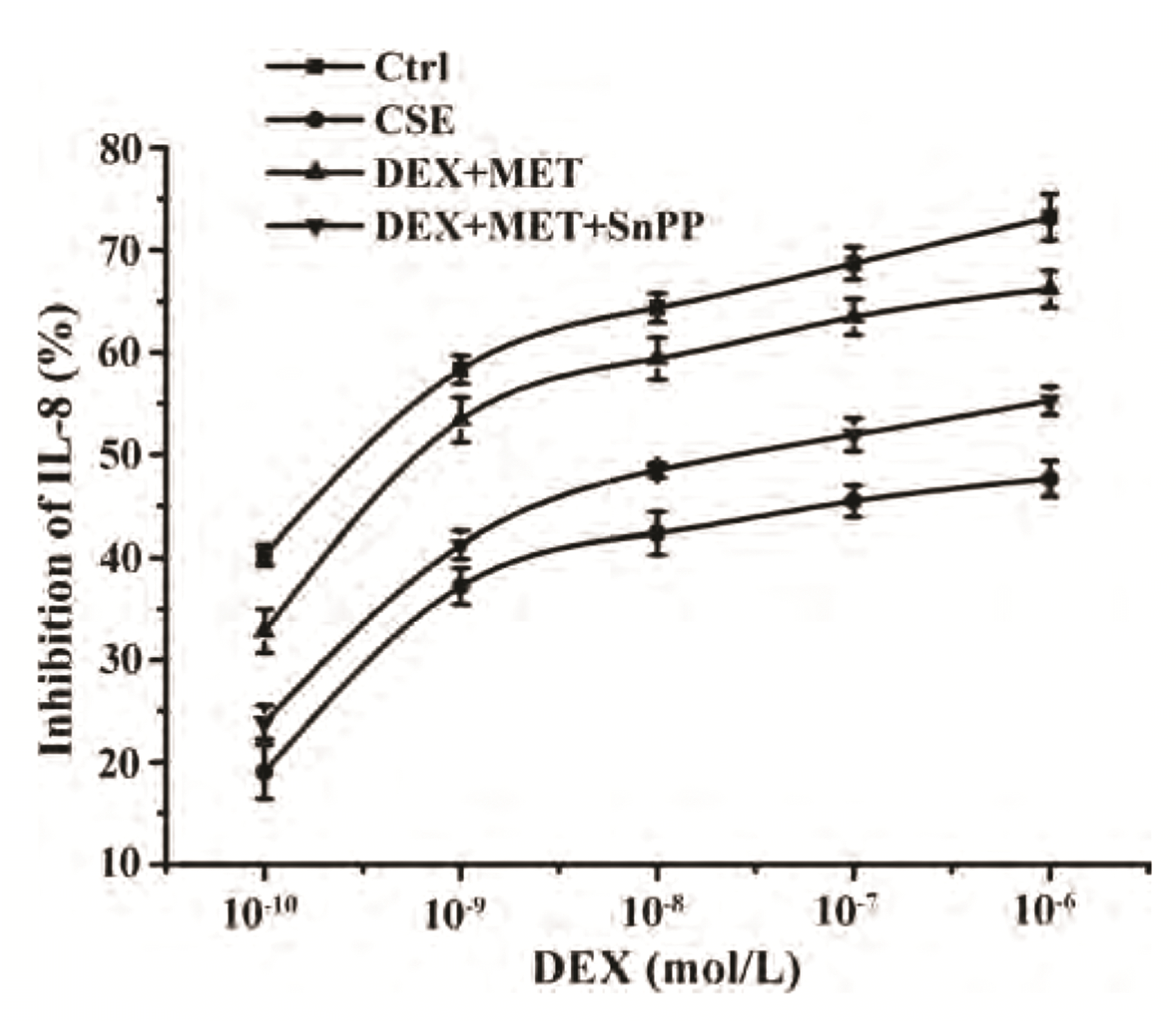 | Fig. 9Effect of SnPP on IL-8 inhibition rates.Data are expressed as means ± SD (n = 3). SnPP, tin protoporphyrin; IL, interleukin; CSE, cigarette smoke extract; DEX, dexamethasone; MET, metformin.
|
Table 3
Comparison of IC50-DEX values of each group (n = 3)
| Group | IC50-DEX |
|---|---|
| Control | (3.93 ± 0.57) × 10–10 |
| CSE | (6.75 ± 3.67) × 10–7* |
| DEX+MET | (2.32 ± 1.29) × 10–9# |
| DEX+MET+SnPP | (6.67 ± 2.76) × 10–8## |
![]()
MK571 weakens the effect of MET on reducing GC resistance
We wished to gain further insight into the role of MRP1 in reducing the GC resistance by MET in 16HBE cells. The latter were administered MK571 (a chemical inhibitor of MRP1 function). IL-8 concentrations in the supernatant were measured by ELISAs to calculate IC50-DEX. Compared with the control group, IC50-DEX of the CSE group was upregulated obviously. IC50-DEX of the combined-treatment group decreased significantly compared with that in the CSE group. Cells pretreated with MK571 inhibited the decline of IC50-DEX compared with that in the combined-treatment group. In summary, the ability of MET to reverse GC resistance could be impaired by the MRP1 inhibitor MK571 (Fig. 10, Table 4).
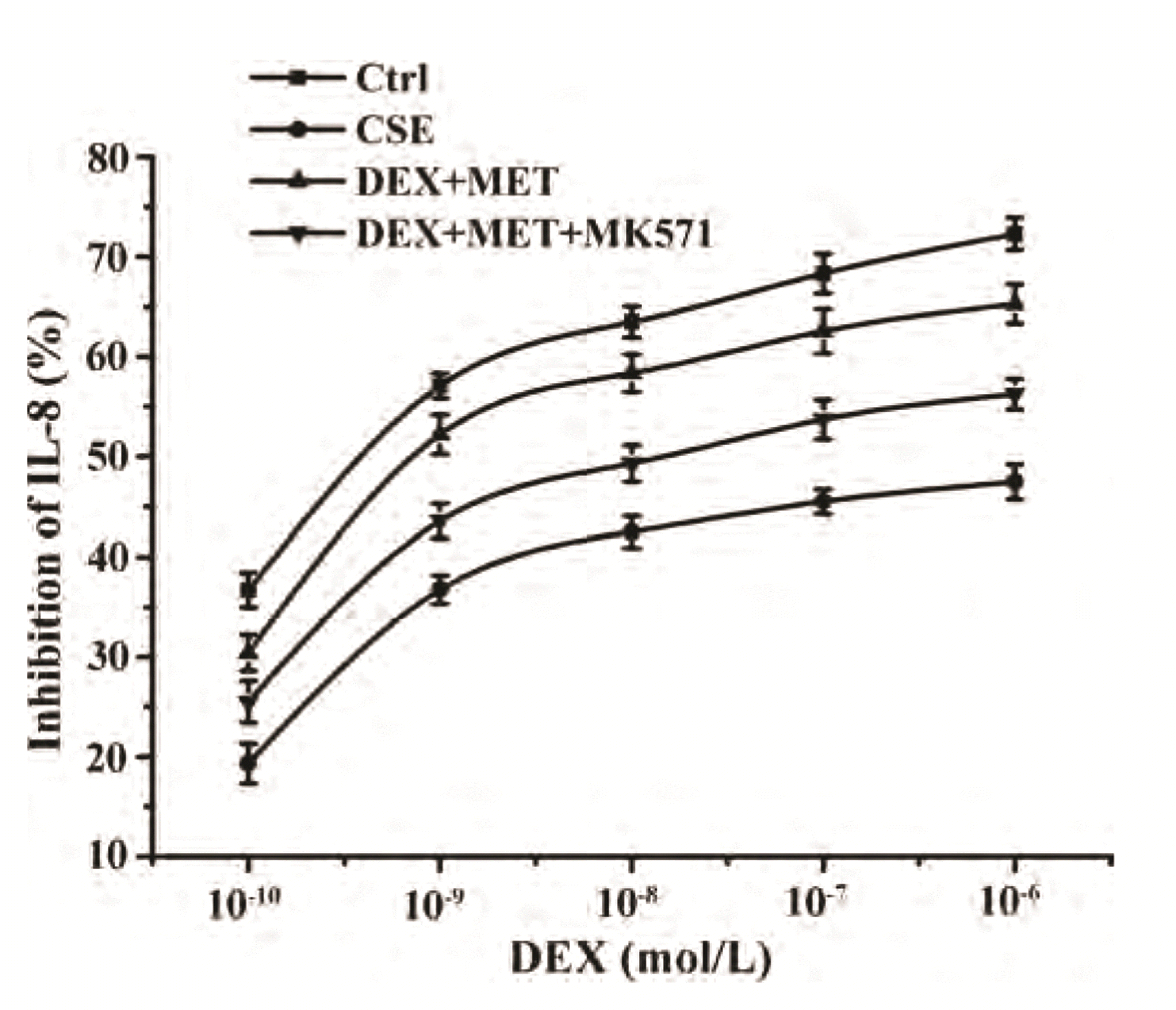 | Fig. 10Effect of MK571 on IL-8 inhibition rates.Data are expressed as means ± SD (n = 3). IL, interleukin; CSE, cigarette smoke extract; DEX, dexamethasone; MET, metformin.
|
Table 4
Comparison of IC50-DEX values of each group (n = 3)
| Group | IC50-DEX |
|---|---|
| Control | (7.11 ± 1.83) × 10–10 |
| CSE | (5.89 ± 1.68) × 10–7* |
| DEX+MET | (3.42 ± 0.95) × 10–9# |
| DEX+MET+MK571 | (4.16 ± 0.81) × 10–8## |
![]()
MK571 decreases the expression of HDAC2
To investigate if MRP1 was involved in regulating HDAC2 expression, MK571 (20 µM) was administered to treated cells. The concentration of an intracellular MRP1 substrate, 5-carboxyfluorescein (5-CF), was measured by flow cytometry to investigate the effect of MET and MK571 on the effusion function of MRP1. HDAC2 expression in each group was also measured by real time-RT-qPCR and western blotting. Compared with the control group, the intracellular fluorescence intensity in the 5-carboxyfluorescein diacetate (5-CFDA) group was increased markedly (Fig. 11), indicating that the experimental model of 5-CFDA effusion had been established. Compared with the 5-CFDA group, CSE stimulation notably increased the intracellular level of 5-CF, whereas combined treatment with MET and DEX reduced the intracellular level of 5-CF distinctly. These results indicated that CSE stimulation inhibited the efflux capacity of MRP1, whereas MET enhanced the efflux capacity of MRP1 significantly, which is consistent with the trend of MRP1 expression described above. The intracellular 5-CF content in the MK571 group was increased compared with that in the combined-treatment group, which indicated that MK571 inhibited MRP1 extravasation. MK571 inhibited expression of the mRNA and protein of HDAC2 significantly compared with that in the combined-treatment group (Fig. 12A–C). In addition, MK571 could reduce expression of HDAC2 protein dose-dependently (Fig. 12D, E). These results showed that inhibition of MRP1 extravasation reduced HDAC2 expression dramatically, which suggested that MRP1 may regulate HDAC2 expression in GC-resistant cells.
 | Fig. 11Effects of MET and MK571 on efflux function of MRP1.(A) Effects of MET and MK571 on 5-CF level. (B) Mean fluorescence intensity (MFI) in each group. Data are expressed as means ± SD. MRP1, multidrug resistance protein 1; MET, metformin; 5-CF, 5-carboxyfluorescein; CSE, cigarette smoke extract; DEX, dexamethasone; 5-CFDA, 5-carboxyfluorescein diacetate. *p < 0.01 vs. control group; **p < 0.01 vs. 5-CFDA group; #p < 0.01 vs. CSE group; Δ p < 0.01 vs. DEX+MET group (n = 5).
|
 | Fig. 12MK571 significantly reduced the expression of HDAC2 protein and mRNA induced by MET.(A) Effect of 20 μM MK571 on the expression of HDAC2 protein. (B) Quantitative analysis results of protein expression in each group. Data are expressed as means ± SD. *p < 0.01 vs. control group; #p < 0.01 vs. CSE group; Δp < 0.01 vs. DEX+MET group (n = 3). (C) Effect of MK571 on mRNA expression of HDAC2. Data are expressed as means ± SD. *p < 0.01 vs. control group; #p < 0.01 vs. CSE group; Δp < 0.01 vs. DEX+MET group (n = 5). (D) Effects of different concentrations of MK571 on the expression of HDAC2 protein. (E) Quantitative analysis results of protein expression in each group. Data are expressed as means ± SD. MET, metformin; HDAC2, histone deacetylase 2; CSE, cigarette smoke extract; DEX, dexamethasone. *p < 0.05 or #p < 0.01 vs. control group (n = 3).
|
Go to :
DISCUSSION
The main characteristics of COPD are inflammation and oxidative stress, which influence each other. Smoking is a high-risk factor for COPD [59]. Various components of tobacco smoke (e.g., heavy metals, nicotinic tar, nicotine) can damage airway epithelial cells directly to initiate inflammatory, antioxidant, and immune mechanisms [60]. The Nrf2-ARE signaling pathway is an important way to regulate inflammation and oxidative stress. For example, curcumin can alleviate airway inflammation and downregulate expression of pro-inflammatory factors in mice by activating the Nrf2 signaling pathway [61]. An activator of Nrf2, bardoxolone methyl, can ameliorate inflammation and oxidative stress in chronic kidney disease [62]. Forsythiaside has been shown to inhibit cigarette smoke-induced lung inflammation by activating Nrf2 and inhibiting NF-κB [63]. Epigallocatechin gallate improves responses to inflammation and oxidative stress in the lungs by activating the Nrf2/Keap1 pathway in rats [64]. Gallic acid, as a natural antioxidant, can improve the redox imbalance in COPD by increasing expression of Nrf2 and its downstream target gene HO-1, and could be used to treat COPD [65].
In COPD, the activity and expression of Nrf2 and its downstream target genes HO-1 and MRP1 are reduced considerably [66]. Simultaneously, the inflammatory response and oxidative stress in the lungs of COPD patients leads to a decrease in the activity and expression of HDAC2 and, eventually, to GC resistance. Studies have shown that HDAC2 activity in the lung parenchyma of patients with COPD is decreased significantly, and that this reduced level is negatively correlated with the concentration of TNF-α and IL-8 [13,26,29]. However, the reasons for the downregulated expression and activity of HDAC2 in COPD are incompletely understood, and may be related to oxidative stress, nitrification stress, and activation of PI3Kδ [67,68]. The excessive production of ROS observed in COPD can lead to excessive oxidation of proteins and lipids. Among them, the level of the lipid-peroxidation product 4-HNE is positively correlated with the occurrence and development of COPD. 4HNE can bind covalently with HDAC2 to destroy protein function, thereby leading to HDAC2 inactivation [22,24,25]. Therefore, anti-inflammation and anti-oxidation strategies are focused upon for the clinical treatment of COPD. Treatment of COPD based on the anti-inflammatory effects of GCs often leads to GC resistance [13]. Thus, finding alternative or complementary therapies for COPD is a rational approach.
The classic hypoglycemic drug MET has been used for treatment of type-2 diabetes mellitus many years. The effect of MET is related to activation of adenosine 5‘-monophosphate-activated protein kinase (AMPK) [69]. Also, increased AMPK activity can reduce inflammation and oxidative stress. It has been found demonstrated that MET can alleviate inflammation and remodeling of the airways by inducing AMPK activation, which provides a new strategy for the treatment of chronic asthma [70]. A recent study showed that MET reduced systemic and pulmonary inflammation in mice exposed to PM2.5, and inhibited the induction and oxidative stress of pulmonary and myocardial fibrosis [71]. MET also has positive effects upon pulmonary disorders (asthma, COPD, lung cancer, lung abscesses), although the mechanism of action is not known, MET proffers a new avenue for COPD treatment [70,72,73].
Most of the commonly used animal models of COPD are induced by exposure to cigarette smoke or direct administration of inflammatory stimulants, (e.g., LPS) [74]. We employed passive smoking combined with tracheal infusion of LPS to establish a COPD model in rats, which is similar to clinical COPD in terms of pathophysiological changes and tissue-structure changes. In addition, 16HBE cells were used as the research object and CSE stimulation was employed as the source of cellular oxidative stress to establish a model of GC resistance in vitro.
Currently recognized methods for evaluating COPD rat models include pulmonary trachea and histopathology, as well as pulmonary function [75]. Compared with the control group, the lung-function indices of the model group (FEV0.3/FVC%, FEF25–75, MMF, PEF) were decreased significantly. Moreover, histopathology of the lung showed that, compared with the control group, the alveolar space in rats in the model group was enlarged irregularly, some alveolar vacuoles were formed, and there was inflammatory-cell infiltration in the interstitium, which resulted in airway stenosis and airway obstruction. The COPD model in rats was established because the pathological and morphological characteristics of lung tissues in the model group were consistent with the pathological and morphological changes of clinical COPD. Upon MET treatment, pulmonary-function indices increased obviously. Histopathology also demonstrated that MET improved alveolar structure and inflammation distinctly. These results indicated that MET could improve the pathological state of COPD.
IL-8 is a mediator of inflammation, and TNF-α is an important chemotactic factor for neutrophils. They promote each other and, eventually, airway remodeling and airflow obstruction occur [76,77]. Notably, studies using animal models of COPD have shown that MET reduces levels of IL-8 and TNF-α, suggesting that MET may improve COPD by inhibiting the inflammatory response. MDA is an important product of lipid peroxidation in cell membranes, whereas SOD and GSH-Px are important members of the antioxidant enzyme system, which reflect the oxidation level in the body. In the MET group, the MDA content in lung tissue decreased, whereas the activities of SOD and GSH-Px increased. Hence, MET may be beneficial to suffering from COPD by having an antioxidant role. These results indicate that MET can improve COPD symptoms through anti-inflammatory and anti-oxidative stress effects.
Under oxidative stress and inflammation, the enhancement of HO-1 expression plays an important part in cell protection. Nardochinoid C can increase HO-1 expression and produce anti-inflammatory and antioxidant effects by activating the Nrf2 signaling pathway, which can be weakened by HO-1 inhibitors [78]. Sodium butyrate reverses the differentiation of T-helper type 17 (Th17) cells into regulatory T cells and attenuates experimental autoimmune uveitis by regulating the Nrf2/HO-1 pathway, whereas SnPP (a HO-1 inhibitor) can eliminate sodium butyrate-mediated inhibition of Th17-cell differentiation and uveitis [79].
MRP1 expression in lung tissue has been shown to play a key part in the resistance to exogenous substances, anti-inflammation effects, and anti-oxidation effects [43]. MRP1 can mediate efflux of the complexes of 4-HNE and GSH, reduce the binding of 4-HNE and HDAC2, maintain HDAC2 activity, and avoid tissue damage [40]. Recent studies have shown that the Nrf2 signaling pathway is not only involved in regulating expression of phase-2 detoxification enzymes, but is also involved in regulating expression of MRPs. Evidence suggests that MRP1 expression is reduced as the Nrf2-ARE pathway is inhibited [43]. Meanwhile, loss of Nrf2 results in a decrease in the HDAC2 level and an increase in lung inflammation [80].
In our animal model of COPD, immunohistochemistry and western blotting showed that CSE stimulation could reduce protein expression of Nrf2, HO-1, MRP1, and HDAC2 in cells, whereas MET could distinctly enhance protein expression of Nrf2, HO-1, MRP1, and HDAC2. The changes observed for Nrf2, MRP1, and HDAC2 were consistent. Therefore, we hypothesized that MET may play an important part in upregulated expression of MRP1 in COPD by activating the Nrf2 signaling pathway. Furthermore, MET could also induce HDAC2 expression in COPD, suggesting that MET may improve GC resistance by upregulating HDAC2 expression because the decrease in HDAC2 expression is a vital reason for GC resistance in COPD. Therefore, we established an in vitro model of GC resistance to explore the effect and mechanism of action of MET on GC resistance.
With respect to the model of GC resistance using 16HBE cells, IL-8 content was measured by ELISAs and IC50-DEX was compared. IC50-DEX was used to illustrate the inhibitory effect of DEX on IL-8 secretion in GC-resistant cells to judge the degree of GC resistance stimulated by CSE. We discovered that CSE stimulation could induce GC-resistant 16HBE cells, and MET could improve the GC resistance of cells, but combined treatment with DEX was more efficacious. However, the HO-1 inhibitor SnPP attenuated the ability of MET to reverse GC resistance. Western blotting showed that the protein expression of Nrf2, HO-1, MRP1, and HDAC2 in the CSE group was reduced markedly, but upregulated notably in the MET group. Also, this effect was more significant when MET was combined with DEX, These outcomes were consistent with the results obtained using in vivo models. Consequently, MET may upregulate MRP1 expression by activating the Nrf2/HO-1 signaling pathway, and further regulate expression of HDAC2 protein to reduce GC resistance.
Next, we administered the MRP1 inhibitor MK571 to investigate if MRP1 is involved in regulating MET to reduce GC resistance. Flow cytometry showed that CSE stimulation inhibited the effect of MRP1, MET could enhance the efflux ability of MRP1 markedly, and MK571 could inhibit upregulation of MRP1 expression by MET. IL-8 levels were measured by ELISAs and IC50-DEX values were compared. We discovered that MK571 weakened the ability of MET to reverse GC resistance. HDAC2 expression was measured by real-time RT-qPCR and western blotting. We found that MK571 inhibited HDAC2 expression dramatically and that expression of MRP1 and HDAC2 was consistent. These data suggested that MRP1 was involved in HDAC2 expression in GC-resistant cells regulated by MET, which regulated GC resistance in COPD. Combined with the experimental results obtained previously, we concluded that MET improves GC resistance by activating the Nrf2/HO-1 signaling pathway, which induces the increased expression and function of MRP1 to further upregulate HDAC2 expression.
In general, MET can improve COPD state by reducing inflammatory response and oxidative stress, and the mechanism may be related to the activation of Nrf2 signaling pathway and its downstream target MRP1. At the same time, MET may enhance the function and expression of MRP1 through activating the Nrf2/HO-1 signaling pathway, thereby promoting the expression of HDAC2, then alleviating the GC resistance. This study provides a new idea for MET in the treatment of glucocorticoid resistance in COPD patients (Fig. 13).
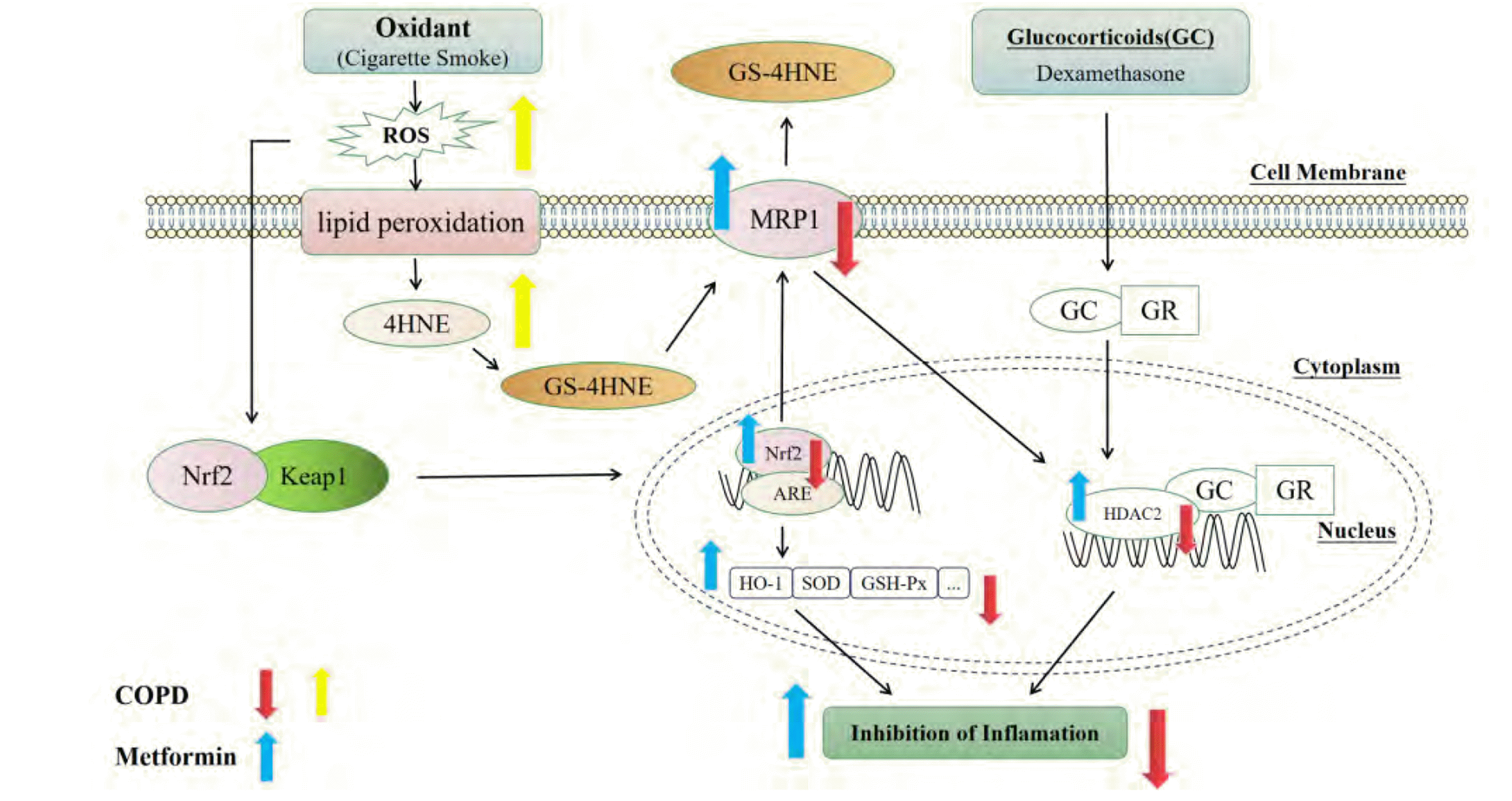 | Fig. 13The potential mechanism of MET alleviating COPD and CSE-induced GC resistance through activating Nrf2/HO-1 signaling pathway, which can promote the function and expression of MRP1 to increase the expression of HDAC2.MET, metformin; COPD, chronic obstructive pulmonary disease; CSE, cigarette smoke extract; GC, glucocorticoid; Nrf2, nuclear factor E2-related factor 2; HO-1, heme oxygenase-1; MRP1, multidrug resistance protein 1; HDAC2, histone deacetylase 2; 4-HNE, 4-hydroxynonenal; ROS, reactive oxygen species; SOD, superoxide dismutase.
|
Go to :
 XML Download
XML Download