

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Rosiglitazone, an oral thiazolidinedione (TZD)-based antidiabetic drug [1,2], is used to reduce blood glucose levels in patients with type 2 diabetes. The original approval of rosiglitazone was based on its effect of reducing the levels of blood glucose and glycated hemoglobin [1,2]. However, it has been reported that rosiglitazone is associated with a significant increase in the risk of myocardial infarction and mortality related to heart diseases [3] and it was once removed from the market due to the possibility of heart toxicity. However, the US Food and Drug Administration (FDA) removed restrictions on the drug's use based on reports that heart toxicity is unlikely and it is still available in the US. In addition, the use of TZD-based diabetes drugs is gradually increasing due to reports that they improve insulin resistance and lower the incidence of cardiovascular disease [4]. Given that more than half of the diabetic patients die from cardiovascular causes, any unexpected effect of antidiabetic therapies on cardiovascular system would be of particular concern. Therefore, extensive research is needed on the various effects that may be caused by rosiglitazone in TZD class on the cardiovascular systems of diabetic patients.
The antidiabetic effect of rosiglitazone is mediated by activation of peroxisome proliferator-activated receptor γ (PPARγ) [1,2]. Besides the PPARγ-mediated antidiabetic action, rosiglitazone also interacts with several types of ion channels in vascular smooth muscle cells. For example, rosiglitazone blocks L-type Ca2+ channels and voltage-gated K+ channels in vascular smooth muscles [5,6] and cloned Kv1.3 and Kv4.3 channels [7,8]. Since rosiglitazone can inhibit voltage-gated K+ channels, we hypothesize here that the adverse cardiovascular effect of rosiglitazone might be mediated by its interaction with Kv1.5 channels (Kv1.5), which is a predominant Kv isoform in the cardiovascular system [9-11].
Kv1.5 channels play an important role in determining the length of cardiac action potentials and, therefore, have been the targets of antiarrhythmic drugs [12]. Because Kv1.5 channels exhibit rapid activation and little inactivation, they can contribute to repolarization of atrial action potentials. Dysfunction of Kv1.5 results in prolongation of cardiac action potentials, which eventually leads to cardiac arrhythmias with serious morbidity [13-15]. Kv1.5 channels underlie the ultrarapid delayed rectifier outward K+ currents, which are present in atrium of heart, but absent from ventricle [14,16].
We here show, with a whole-cell patch-clamp technique, that rosiglitazone acts as an open channel blocker of Kv1.5 channels expressed in Chinese hamster ovary (CHO) cells. As the Kv1.5 channels play a crucial role in controlling the duration of cardiac action potentials and hence the atrial rhythm, our data imply that rosiglitazone may interfere with the cardiac rhythm.
Go to : 
METHODS
Cell culture and transfection
CHO cells derived from rat brain were used for Kv1.5 expression and electrophysiological recordings [17]. Kv1.5 cDNA [16] was transferred into the plasmid expression vector pCR3.1 (Invitrogen Corporation, San Diego, CA, USA). CHO cells were transfected with Kv1.5 cDNA using FuGENE 6 reagent (Boehringer Mannheim, Indianapolis, IN, USA). The transfected cells were cultured in Iscove’s modified Dulbecco’s medium (Invitrogen Corporation) supplemented with 10% fetal bovine serum, 2 mM glutamine, 0.1 mM hypoxanthine, 0.01 mM thymidine, and 300 µg/ml G418 (A.G. Scientific, San Diego, CA, USA). The cultures, incubated in 95% humidified air-5% CO2 at 37°C, were passaged every 4–5 days with a brief trypsin-EDTA treatment followed by seeding onto glass coverslips (diameter: 12 mm; Fisher Scientific, Pittsburgh, PA, USA) in a Petri dish. The cells were used for electrophysiology 12–24 h after the seeding.
Electrophysiology
Kv1.5 currents were recorded from CHO cells, with a whole-cell patch-clamp technique [18] at 22–23°C. The micropipettes were fabricated from glass capillary tubing (PG10165-4; World Precision Instruments, Sarasota, FL, USA) with a double-stage vertical puller (PC-10; Narishige, Tokyo, Japan), and had a tip resistance of 2–3 MΩ when filled with a pipette solution composed of 140 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 10 mM EGTA (pH 7.3 with KOH). Whole-cell currents were amplified with Axopatch 200B amplifier (Molecular Devices, San Jose, CA, USA), digitized with Digidata 1440A (Molecular Devices) at 5 kHz and low-pass filtered with a four-pole Bessel filter at 2 kHz. Pipette and whole-cell capacitive currents were canceled and series resistance was compensated at 80% with the amplifier, while leak subtraction was not used. Generation of voltage commands and acquisition of data were controlled with pClamp 10.1 software (Molecular Devices). Recording chamber (RC-13; Warner Instrument Corporation, Hamden, CT, USA) was continuously perfused at 1 ml/min with a bath solution: 140 mM NaCl, 5 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 20 mM HEPES and 10 mM glucose (pH 7.3 with NaOH). The vehicle for rosiglitazone (Cayman Chemical Co., Ann Arbor, MI, USA) was < 0.1% of dimethyl sulfoxide (DMSO), which itself had no effect on Kv1.5 currents (data not shown).
Data analysis
Data were analyzed with Origin 7.0 (OriginLab Corp., Northampton, MA, USA) and Clampfit 10.1 software (Molecular Devices). A model of interaction kinetics between drug and channel was based on a first-order blocking scheme as previously described [19]. This scheme allowed us to obtain IC50 and Hill coefficient (n) from a dose-response curve based on the following equation:
in which I (%) is the percent inhibition of current (I [%] = [1 – Idrug/Icontrol] × 100) and [D] represents various drug concentrations. The steady-state activation curve was fitted with the Boltzmann equation:
where k is the slope factor, V is the test potential, and V1/2 is the potential at which the conductance was half-maximal. We obtained an activation time constant by fitting the latter 50% of activation (i.e., rise from 50% to 100% of peak amplitude) with a single exponential function. Since the drug-channel interaction was based on the first-order scheme, binding (k+1) and unbinding (k–1) rate constants were obtained from the following equations:
in which τD is the drug-induced time constant. The drug-induced time constant and deactivation time constant were determined by fitting with the sum of the exponentials:
where τ1, τ2, and τn are the time constants; A1, A2, and An are the amplitudes of each component of the exponential; and B is the baseline value.
Results were expressed as means ± SEM. Student’s t-test and analysis of variance (ANOVA) were used for statistical analysis with a confidence level of p < 0.05.
Go to :
RESULTS
Inhibition of Kv1.5 by rosiglitazone
We examined the effects of rosiglitazone on potassium currents mediated by Kv1.5 channels, using a whole-cell patch-clamp technique. Whole-cell currents were elicited with 250-ms depolarizations to +50 mV in CHO cells expressing Kv1.5 channels. The currents showed typical characteristics of Kv1.5: rapid activation and very slow inactivation (Fig. 1A) [17]. Bath-applied rosiglitazone reduced the Kv1.5 current amplitudes and accelerated its decay rate in a concentration-dependent manner (Fig. 1A). The suppression of Kv1.5 currents for each concentration of rosiglitazone reached a steady state within 2 min. The rosiglitazone-mediated suppression of end-pulse currents was more pronounced than that of peak currents. A nonlinear least-squares fit of dose-response plots with the Hill equation yielded an IC50 value of 18.9 ± 1.9 µM and a Hill coefficient of 1.8 ± 0.2 (n = 5) for the end-pulse currents, and an IC50 value of 65.2 ± 3.2 µM and a Hill coefficient of 1.6 ± 0.2 (n = 5) for the peak currents (Fig. 1B). In subsequent experiments, 20 µM rosiglitazone, slightly higher than the IC50, was used. The effect of rosiglitazone was reversible: the suppression of end-pulse currents caused by 20 µM rosiglitazone was reversed to 84.4 ± 3.4% of the pre-drug baseline (n = 5) after 2-min wash-out (Fig. 1C).
 | Fig. 1Concentration dependence of rosiglitazone-induced inhibition of Kv1.5 currents.(A) Kv1.5 currents were elicited with +50 mV depolarization (250 ms) from a holding potential of –80 mV, every 10 sec. Currents recorded in the absence and presence of 3, 10, 30, and 100 µM rosiglitazone were superimposed. The dotted line represents zero current. (B) Dose response curve of rosiglitazone-mediated reduction in Kv1.5 current. The amplitudes of Kv1.5 current were measured at the end (closed circle) and peak (open circle) of the depolarizing pulses at various concentrations of rosiglitazone. The data of % inhibition (I [%] = [1 – Irosiglitazone/Icontrol] × 100) were fitted with the Hill equation (solid lines). (C) Time course of Kv1.5 inhibition by rosiglitazone. The current amplitudes were measured at the end of a 250-ms depolarizing pulse and normalized to the baseline amplitude. Data are expressed as mean ± SEM.
|
The larger effect of rosiglitazone on end-pulse current than on peak current implies that rosiglitazone might block Kv1.5 channels only after they are open [17]. If rosiglitazone acts as an open channel blocker of Kv1.5, it is predicted that the activation time constant would be resistant to rosiglitazone. Indeed, the activation time constant of Kv1.5 currents, elicited with a 250-ms depolarizing pulse from –80 to +50 mV, was not significantly affected by rosiglitazone: 1.7 ± 0.17 ms (n = 5) in control and 1.6 ± 0.14 ms (n = 5) in 30 µM rosiglitazone. Based on the difference in the rosiglitazone-mediated suppression between end-pulse and peak currents, and the ineffectiveness of rosiglitazone on the activation time constant, we hypothesized that rosiglitazone might inhibit Kv1.5 current as an open channel blocker.
Voltage-dependence of rosiglitazone-mediated inhibition of Kv1.5
We next investigated whether the inhibition of Kv1.5 currents caused by rosiglitazone was dependent on membrane potential. If rosiglitazone blocks Kv1.5 channels that are in an open state, the rosiglitazone-mediated inhibition would emerge in a voltage range where the channels begin to be activated, and then remain constant once they are fully activated. To test this prediction, we constructed a current-voltage (I-V) relationship in the absence and presence of 20 µM rosiglitazone (Fig. 2A–C). Outward Kv1.5 currents began to be generated at –30 mV and linearly increased with higher depolarizing pulses (Fig. 2A, C). When channel conductance was calculated from the I-V relationship, it steeply increased in the voltage range from –30 to 0 mV and remained in a fully active state at positive potentials (Fig. 2D, dotted line). The I-V relationship showed a sigmoidal shape at potentials between –30 and 0 mV. Rosiglitazone (20 µM) reduced Kv1.5 currents in the whole voltage range over which Kv1.5 was activated, i.e., from –30 to +40 mV (Fig. 2B, C).
 | Fig. 2Voltage dependence of rosiglitazone-induced inhibition of Kv1.5 currents.The Kv1.5 currents were produced by applying 250-ms depolarizing pulses between –60 and +40 mV in 10-mV increments every 10 sec from a holding potential of –80 mV under control conditions (A), and after the addition of 20 µM rosiglitazone (B). The dotted lines in (A) and (B) represent zero current. (C) The amplitudes of Kv1.5 currents were measured at the end of test pulses and plotted against membrane potentials, in control (○) and 20 µM rosiglitazone (●). (D) The activation curve of control Kv1.5 current was constructed from tail current amplitudes at –40 mV after 250-ms depolarizing pulses between –60 and +40 mV in 10-mV. Only the Boltzmann fitting curve is shown (dotted line, normalized current y-axis for activation curve). The percent inhibition of Kv1.5 current by rosiglitazone was plotted (●) for comparison with the activation profile. In the voltage range between –20 and 0 mV, the percent inhibition increased steeply suggesting an open channel blockade. The solid line was drawn from a linear curve fitting the relative current data between 0 and +40 mV. Data are expressed as mean ± SEM.
|
To examine the voltage-dependence of the rosiglitazone effect, we plotted percent inhibition of Kv1.5 currents (see Methods) against membrane potential (Fig. 2D). The degree of inhibition varied with voltage between –20 and 0 mV, which is the range of gradual channel opening (Fig. 2D): 34.5 ± 3.8% inhibition at –20 mV and 63.3 ± 3.2% inhibition at 0 mV (n = 4, p < 0.05). However, the inhibition of Kv1.5 by rosiglitazone at potentials between 0 and +40 mV, where the channels are fully activated, lacked such voltage dependence: 63.3 ± 3.8% inhibition at 0 mV and 64.3 ± 2.2% inhibition at +40 mV (n = 4, ANOVA, p < 0.05). As a result, the linear curve fitting of the data at positive potentials (Fig. 2D, solid line) showed a slope of zero. In sum, the voltage-dependence of the effect of rosiglitazone implies that the inhibition of Kv1.5 occurs preferentially after the channels are open. Furthermore, the data indicate that, once channels are fully activated, the effect of rosiglitazone is independent of membrane potentials.
Decay kinetics of Kv1.5 currents in rosiglitazone
If the interaction between drug and channel is a first-order reaction, a KD value calculated from binding (k+1) and unbinding (k–1) coefficients would be similar to the KD obtained from an empirical dose-response curve. Furthermore, in a first-order reaction, the binding and unbinding coefficients could be obtained from a decay time constant (τD) of currents (see Methods, equation 3). As shown in Fig. 3A, rosiglitazone accelerated the decay of Kv1.5 current in a concentration-dependent manner. At 10, 30, and 100 µM of rosiglitazone, the current decay at +50 mV was well fitted with a single exponential function. We excluded the data with 3 µM rosiglitazone because the intrinsic slow inactivation of the Kv1.5 current could not be distinguished from the rosiglitazone-induced acceleration. The plot of 1/τD at +50 mV against the rosiglitazone concentrations showed a linear relationship (Fig. 3B). A linear fit of the plot yielded a binding rate constant (k+1) of 0.27 ± 0.04 µM–1s–1 and an unbinding rate constant (k–1) of 3.88 ± 0.13 s–1 (n = 5). From these two constants, we derived a theoretical KD value (k–1/k+1) of 14.4 µM, which is close to the experimental IC50 value of 18.9 µM obtained from the concentration-response curve in Fig. 1.
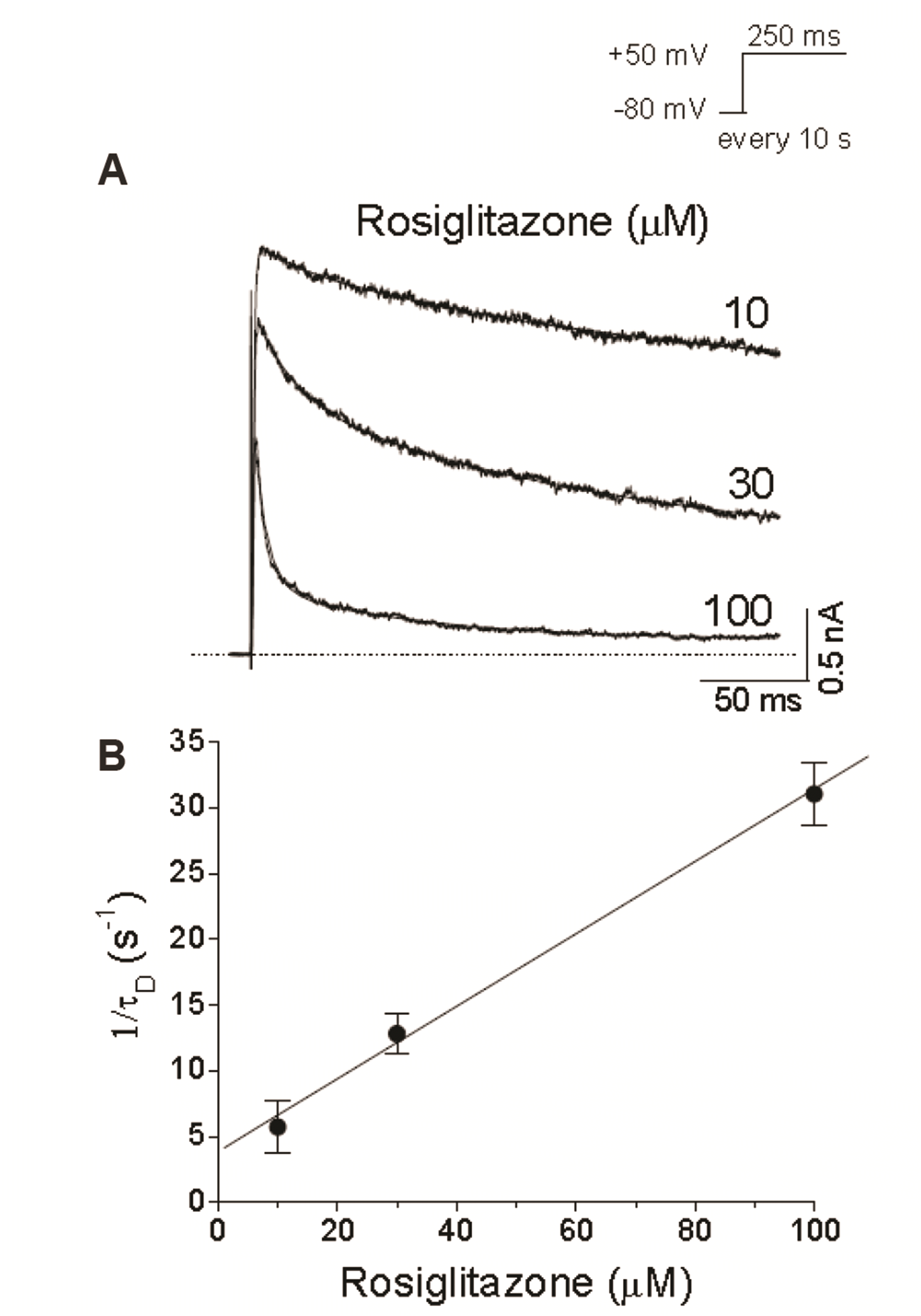 | Fig. 3Concentration-dependent kinetics of Kv1.5 inhibition caused by rosiglitazone.(A) Kv1.5 currents were elicited with +50 mV pulses (250 ms) every 10 sec in the presence of rosiglitazone (10, 30, and 100 µM). The dotted line represents zero current. (B) A drug-induced time constant (τD) was obtained from a single exponential fitting to the decaying phase of Kv1.5 current. The inverse of τD obtained at +50 mV was plotted against rosiglitazone concentrations. The solid line represents linear fit of the data. A binding rate constant (k+1) and an unbinding rate constant (k–1) were obtained from the slope and intercept values of the fitted line, 1/τD = k+1[D] + k–1. Data are expressed as mean ± SEM.
|
Deactivation kinetics
Deactivation kinetics can provide useful information about open channel blockers. If a blocker binds to channels in their open state, channel closing would be slowed down compared to the closing speed of drug-free channels [7,17,20,21]. We used tail currents of Kv1.5 to investigate the deactivation property of Kv1.5 and the effect of rosiglitazone on its kinetics. Fig. 4 shows superimposed representative tail currents recorded with 250-ms repolarizing pulse of –40 mV after a 250-ms depolarizing pulse of +50 mV from a holding potential of –80 mV, under control conditions and in the presence of 20 µM rosiglitazone (Fig. 4). Rosiglitazone (20 µM) reduced the initial amplitude of tail current and slowed the decay time course of tail currents. When fitted with a single exponential function, control tail currents declined with a time constant of 23.2 ± 2.7 ms (n = 4) at –40 mV. In the presence of 20 µM rosiglitazone, the decay time constant (32.8 ± 3.0 ms) was larger than the control one (Student’s t-test, p < 0.05). Since rosiglitazone reduced the initial amplitude of tail currents and slowed the decay kinetics, an overlap of two tail currents in the absence and presence of rosiglitazone revealed a “tail crossover” phenomenon (Fig. 4).
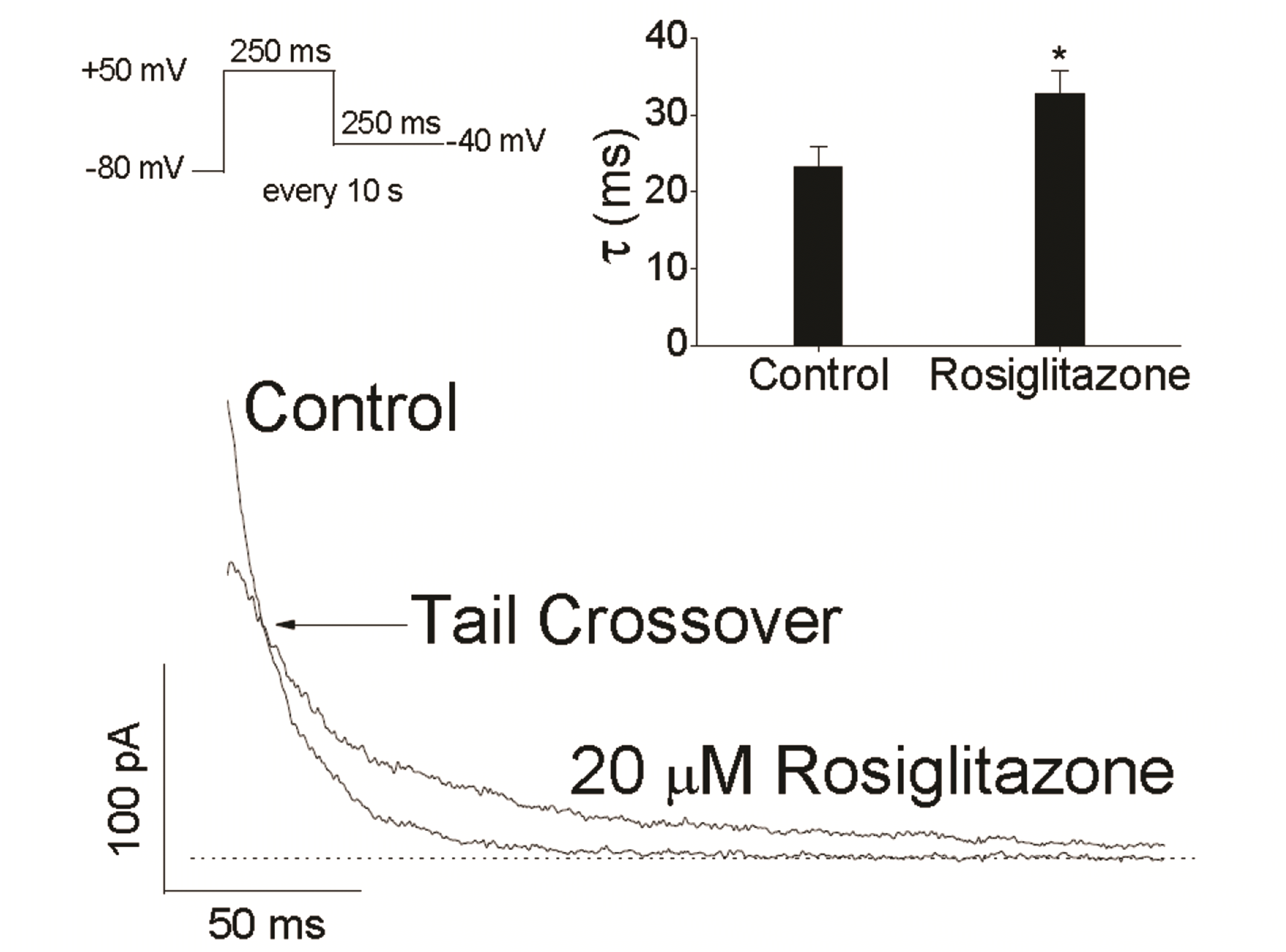 | Fig. 4Effects of rosiglitazone on the deactivation kinetics of Kv1.5 currents.Tail currents were induced with the repolarizing pulse of –40 mV after a 250-ms depolarizing pulse of +50 mV, in the absence and presence of 20 µM rosiglitazone. Superimposition of two tail currents revealed tail crossover (arrow). The dotted line represents zero current. Inset, mean time constants of the tail current decay in the presence of rosiglitazone was significantly larger than that in control condition (Student’s t-test, n = 4; *p < 0.05).
|
Use-dependent inhibition of Kv1.5 by rosiglitazone
Open channel blockers typically display use-dependent inhibition because the blockers would have a higher chance to bind to channel pores as the channels open more frequently. To test for the use dependence of rosiglitazone-mediated inhibition, we activated Kv1.5 currents with a 125-ms depolarizing pulse (+50 mV) from a holding potential of –80 mV, 10 times at 1 Hz and 20 times at 2 Hz, respectively. For the two different stimuli, the duration of stimulation is equal to 10 sec. After 2-min exposure to 20 µM rosiglitazone at –80 mV (i.e., without any depolarization), the 1- and 2-Hz depolarization protocols resumed. In the absence of rosiglitazone, the progressive suppression of Kv1.5 current was marginal during the 1- and 2-Hz stimuli (Fig. 5A). The peak amplitude of the 10th or 20th Kv1.5 current slightly decreased from the first current by 5.7 ± 0.9% (n = 4) at 1 Hz and by 15.1 ± 4.1%] (n = 4) at 2 Hz. However, in the presence of 20 µM rosiglitazone, the peak amplitude of Kv1.5 current was gradually reduced by 45.4 ± 4.9% (n = 4) at the 10th stimulus delivered at 1 Hz, and by 65.4 ± 4.3% (n = 4) at the 20th stimulus delivered at 2 Hz (Fig. 5A), indicating robust use-dependent blockade by rosiglitazone. Interestingly, without any activation of Kv1 .5 channels, 20 µM rosiglitazone itself had little effect on the current amplitude as the first current in a stimulus set was not significantly different between control and rosiglitazone conditions. This also strongly indicates that rosiglitazone may not bind to Kv1.5 channels that are in closed state.
 | Fig. 5Use-dependence of rosiglitazone-mediated inhibition of Kv1.5.(A) Superimposed Kv1.5 current traces elicited successively with depolarizing pulses (+50 mV, 125 ms) at 1 or 2 Hz. The dotted lines represent zero current. Dashed lines are for a comparison of the peak amplitudes of the first currents in both stimuli sets. (B) Plot of normalized amplitude of peak current 1 (circles) and 2 Hz (triangles), in control (open symbols) and in 20 µM rosiglitazone (closed symbols), against the elapsed time axis. The peak amplitudes were normalized to the peak amplitude of the first current in each condition. (C) With the data in (B), the current amplitude in rosiglitazone was normalized to the control amplitude at each elapsed time. Data are expressed as mean ± SEM.
|
If rosiglitazone is a use-dependent blocker of Kv1.5, the magnitude of blockade would increase with the degree of use, or channel opening. In other words, it is predicted that the use-dependent blockade at 2 Hz stimuli will be larger than that at 1 Hz stimuli. The suppression by 2 Hz stimuli indeed appeared to be larger than that by 1 Hz in rosiglitazone (Fig. 5A, B), but the progressive reduction of the control current by repetitive stimuli obscured accurate determination of the comparison between 1- and 2-Hz stimuli. Therefore, we normalized the currents in rosiglitazone to the corresponding control currents in a set of stimuli (Fig. 5C). The plot of the normalized current against the elapsed time revealed a larger blockade at 2 Hz, further confirming the use-dependence of rosiglitazone.
Effects of rosiglitazone on the kinetics of Kv1.5 recovery from inactivation
Slow inactivation, as shown in the control current trace of Kv1.5 (Fig. 1), is a prominent feature of the Kv1.5 channels. Therefore, an estimation of time required for recovery from inactivation may be necessary for comprehensive understanding of the reaction scheme of Kv1.5 channels. In addition, an analysis of the effect of rosiglitazone on the recovery time can provide the information how the drug alters the inactivation process and/or state. The degree of recovery from inactivation was measured with a two-pulse protocol, where two depolarization pulses (250 ms, +40 mV) were given in succession with various intervals (Fig. 6A). If some of the channels that are activated by the first pulse still remain inactivated until the second pulse is given, the amplitude of the second current would be smaller than that of the first one.
 | Fig. 6Effects of rosiglitazone on the kinetics of Kv1.5 recovery from steady-state inactivation.(A) The degree of recovery was assayed with the following two-pulse protocol: the first prepulse (+40 mV, 250 ms) was followed by a second identical pulse with a varying interpulse interval ranging from 5 to 10,000 ms. The pair of pulses was applied at an interval of 30 sec. (B) The peak amplitude of the second current in the pair (■ in A) was normalized to the peak of the first current (□ in A), and plotted against interpulse intervals. (C) The range of y-axis values in (B) were expanded to the range from 0 to 1 by normalizing the minimum values in (B) (at 5 ms interval) to 0. This re-normalization revealed faster recovery from inactivation in rosiglitazone than in control condition. The curves in (B and C) were obtained from a double exponential fitting, from which two recovery time constants were obtained. (D) Summarized data of the fast and slow time constants obtained from the double exponential fittings (n = 4; *p < 0.05 vs. data under control conditions). Data are expressed as means ± SEM.
|
We estimated the degree of recovery by plotting the ratio of “the second peak current/the first peak current” against the duration of inter-pulse interval (Fig. 6B, C). This plot was well fitted with a double exponential function. In the control condition, the time constants of fast and slow components were 388.2 ± 46.5 ms and 3,851.2 ± 582.7 ms, respectively (n = 4). Rosiglitazone (20 µM) significantly decreased the fast time constant to 199.8 ± 17.2 ms (p < 0.05, n = 4) whereas it did not alter the slow component (3,924.9 ± 276.4 ms). This result indicates that one component of the recovery processes of Kv1.5 was accelerated by rosiglitazone. If the inactive state or “non-conducting state” of Kv1.5 in the presence of rosiglitazone is qualitatively indistinguishable from that under control condition, the recovery time constants would not be affected by rosiglitazone. Therefore, the rosiglitazone-induced abbreviation of time constant implies that the non-conducting, resting state in rosiglitazone might be different from the inactive state under control condition. This aspect will be further discussed.
Go to :
DISCUSSION
The present study shows that rosiglitazone, an antidiabetic drug, blocks Kv1.5 channels expressed in CHO cells. The rosiglitazone-mediated inhibition of Kv1.5 is characterized by a concentration-dependent acceleration of the apparent rate of current decay. These results are similar to those presented with various open channel blockers [17,20-25]. The characteristics of the rosiglitazone-induced inhibition of Kv1.5 suggest that rosiglitazone preferentially interacts with the open state of Kv1.5 channels based on the following lines of evidence. 1) Rosiglitazone accelerated the rate of Kv1.5 current decay during a depolarizing pulse (Fig. 1A). 2) At the onset of depolarizing pulses, rosiglitazone had no effect on the initial time course of channel activation indicating that rosiglitazone does not bind to the closed or resting state of Kv1.5 (Fig. 1D). 3) Rosiglitazone did not reduce Kv1.5 currents when the channels remain closed (Fig. 5A). The absence of tonic blockade by rosiglitazone suggests that the drug does not interact with the Kv1.5 in the closed or resting state. 4) The inhibition induced by rosiglitazone was voltage-dependent and increased steeply in the voltage range of channel activation (Fig. 2D). 5) Rosiglitazone slowed the deactivation time course, resulting in a tail crossover phenomenon (Fig. 4). This latter phenomenon suggests an interaction between rosiglitazone and the open state of Kv1.5 [7,17,20,21]. The inhibition of Kv1.5 channels by rosiglitazone is use-dependent and enhanced by higher rates of channel activation. This is consistent with the actions of rosiglitazone on the open state of Kv1.5 [17,20].
Drugs that interact predominantly with open state of the channel can do so by moving into the ion-conducting pore. The inhibition produced by rosiglitazone was voltage-dependent and increased steeply between 0 and +30 mV, which correspond to the voltage range for channel opening. However, the block of Kv1.5 by rosiglitazone was voltage-independent in the positive voltage range despite Kv1.5 being fully activated at this voltage range. The pKa values of rosiglitazone are 6.1 and 6.8 [26] and it is predominately uncharged at the intracellular pH of 7.3 (pH of the pipette solution). Therefore, no additional block was detected in the voltage range where channels are fully activated (Fig. 2), suggesting that the interaction between rosiglitazone and Kv1.5 is independent of the transmembrane electric field.
In our study, rosiglitazone accelerated the kinetics of the recovery from inactivation process (Fig. 6). It was reported that, in the reaction of open channel blockade, the recovery from inactivation is slowed by the channel blocker because the transition of OR ↔ O ↔ C in the presence of drug is slower than the transition of O ↔ C in control conditions [17,20], where OR is a drug-bound open state, O is an open state and C is a closed state. The reason for the discrepancy between our and the previous studies is unclear, but it is possible that rosiglitazone may leave the channel when the inactivation gate is open, whereas other drugs leave the channels only in open state. Once bound to an open channel, rosiglitazone can be trapped in the channel, and the consequent structural change of the channel may accelerate the opening of the inactivation gate or shorten the dwelling time in inactivated state. Further elucidation of the mechanism will be of future interest.
TZD-based drugs, including rosiglitazone, are good drugs that improve diabetes by directly activating PPARγ and improving insulin resistance [1,2,27-29]. However, these drugs are restricted in their use due to concerns over side effects related to heart toxicity. As a representative example, rosiglitazone, a TZD-based diabetes treatment, has been withdrawn from the market in most countries, including Europe. However, the US FDA did not agree on market eviction, and a survey conducted after the issue of market eviction reported that side effects on the heart were low compared to other diabetes treatments, which lifted restrictions and is currently used through strict prescriptions in the US. However, since TZD-based drugs show excellent effects on improving insulin resistance [4], they are very effective in treating type 2 diabetes patients, which is likely to increase the use of TZD-based drugs. Therefore, it is of great significance to examine the side effects or beneficial effects of the heart that may accompany rosiglitazone when administered to type 2 diabetes patients. The results of this study show that rosiglitazone inhibits Kv1.5. Reduction in Kv1.5 currents can induce prolongation of cardiac action potentials [30] resulting in detrimental alteration in cardiac spike. Therefore, it is possible the aforementioned side effects of rosiglitazone might be caused, at least in part, by suppression of Kv1.5 channel in heart.
On the basis of pharmacokinetics of rosiglitazone in patients suffering from type 2 diabetes, the therapeutic human plasma concentrations of rosiglitazone are reported to vary between 0.3 and 2 µM in human patients with diabetes [2,31,32]. The concentration in this range is the plasma concentration measured when rosiglitazone is orally administered to diabetic patients. Therefore, the inhibition of Kv1.5 by rosiglitazone measured in the results of this study may not cause cardiac side effects at the therapeutic level. However, although this plasma level is lower than the IC50 value of rosiglitazone in this study, the difference in membrane phospholipid composition between native cardiac cells and CHO cells may affect the efficacy of rosiglitazone. Furthermore, drug concentrations in tissues may be higher than in plasma due to its high lipophilicity and affinity for adipose tissues. All of these unique properties of in vivo cells may limit the direct extrapolation of our data to clinical settings. Nevertheless, our study clearly demonstrates the possibility of cardiac side effects of rosiglitazone, and thus, raises a concern of using the drug for diabetic patients with cardiac complications.
In conclusion, the present study has described, for the first time, the suppressive effects of rosiglitazone on the Kv1.5 expressed in CHO cells. Detailed study of the interaction kinetics between rosiglitazone and Kv1.5 suggests that rosiglitazone is an open-channel blocker for Kv1.5 in a concentration-, voltage-, time-, and use-dependent manner. This study reveals an unappreciated action of the antidiabetic drug, and therefore, stresses the need for further investigation of the effect of rosiglitazone on cardiac function in vivo.
Go to :
 XML Download
XML Download