

 PDF
PDF Citation
Citation Print
Print



Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a rare, rapidly progressive, and life-threatening hematologic disorder with excessive immune activation [1]. HLH is a group of disorders characterized by accumulation of lymphocytes and macrophages leading to ‘hemophagocytosis’ (phagocytosis of hematologic cells by macrophages) [2]. If HLH is left untreated, most patients survive for only a few months with severe progressive multiple organ failure [1]. In 1983, the long-term survival in patients with HLH was reportedly only 4% [3]. HLH can be divided into two major categories; primary and secondary [4]. Primary HLH includes familial HLH (FHL), X-linked lymphoproliferative disease type 1 (XLP1), and other primary immunodeficiencies associated with HLH and partial albinism [5,6]. However, secondary HLH can develop because of various triggers, including infections, neoplasms, and autoimmune conditions [4]. HLH most frequently affects infants aged 0 to 18 months; however, the condition is also observed in children, adolescents, and adults of all ages [7].
In 1991, the Histiocyte Society first presented the diagnostic guidelines for HLH based on the common clinical, laboratory, and histopathological findings of HLH [8]. Subsequently, in 1994, the prospective international treatment protocol, HLH-94, based on etoposide, corticosteroids, and cyclosporine A (CSA), was introduced [9]. The HLH-94 protocol dramatically increased the survival rate of patients with HLH to 54% with a median 6-year follow-up [1,10]. Thereafter, a modified treatment protocol, HLH-2004, with revised diagnostic criteria, was introduced in 2004 [11]. In HLH-2004, CSA was incorporated into induction therapy; however, the survival outcomes of patients with HLH were not different between the two studies [11,12]. Although secondary HLH is more common, both HLH-94 and HLH-2004 protocols mainly focused on pediatric patients with primary HLH [1]. Therefore, implementing the same treatment regimen in all HLH subtypes has been questioned [13].
Although primary and secondary HLH exhibit common clinical features in the beginning, the distribution of each subtype and their respective prognoses vary globally [14,15]. According to a nationwide study on pediatric patients with HLH by the Korea Histiocytosis Working Party from the Korean Society of Hematology, the most common genetic cause of primary HLH was FHL type 3 with UNC13D variants [13]. Moreover, secondary HLH induced by Epstein-Barr virus (EBV) infection had a relatively high incidence, and the 5-year overall survival (OS) rate was 68% in 251 Korean children with HLH [13]. According to the reports on HLH in adults, neoplasms as triggers are more common in adults than in children [16]. Additionally, secondary HLH is the most common HLH subtype in adults [17]. However, to date, there have been no data regarding HLH in adult patients or a comparison of HLH in pediatric and adult patients in Korea. Therefore, we aimed to retrospectively investigate the causes and characteristics of HLH in both pediatric and adult patients with different HLH subtypes in this study. We also investigated the survival rates of and risk factors for poor prognosis in children and adults with HLH.
Go to : 
Materials and methods
1. Subjects and ethical statement
In this study, patients diagnosed with HLH at Keimyung University Dongsan Hospital and Yeungnam University Medical Center in Daegu, Korea between January 2001 and December 2019 were investigated. The medical records of patients with HLH were retrospectively reviewed for age at diagnosis, sex, HLH etiology, treatment regimen, and death.
2. Definition and methods
HLH was diagnosed according to the diagnostic criteria presented by the Histiocyte Society in 1991 and updated in 2004 [8,11]. According to the HLH-2004 guideline, at least five of the eight listed items must be met for the diagnosis of HLH: (1) fever ≥38.5°C; (2) splenomegaly; (3) bicytopenia affecting ≥2 cell lines (hemoglobin <90 g/L [hemoglobin <100 g/L in infants <4 weeks], platelet <100×109/L, and neutrophil <1.0×109/L); (4) hyperferritinemia (ferritin >500 μg/L); (5) hypertriglyceridemia (fasting triglyceride >3.0 mmol/L [265 mg/dL]) and/or hyperfibrinogenemia (fibrinogen, <1.5 g/L); (6) hemophagocytosis in the bone marrow (BM), lymph node, spleen, or liver without evidence of malignancy; (7) elevated levels of soluble CD25 (also called interleukin-2 receptor) >2,400 U/mL; and (8) low or absent natural killer (NK) cell activity [5,11]. In this study, NK cell activity could not be tested using flow cytometry; instead, interferon-γ was measured by enzyme-linked immunosorbent assay using the NK Vue kit (ATgen, Seongnam, Korea) through Seoul Clinical Laboratories (Yongin, Korea; http://www.scllab.co.kr) [18]. Since this external test method was not performed daily, the results of this test were available after several days. Therefore, it was not used directly as a diagnostic criterion but was referenced in previously diagnosed HLH patients. Additionally, mutations in the genes of typical FHL (PRF1, UNC13D, STX11, STXBP2), XLP1 (SH2D1A), and XLP2 (BIRC4) are sufficient to establish a diagnosis of HLH regardless of the number of fulfilled criteria of HLH-2004 [5,6,11].
3. Statistical analysis
The variables are described as median values and ranges. The Mann-Whitney U-test was used to compare the variables between the two groups. The chi-square test was used to compare the ratio between the two groups. The 5-year OS rates between the two groups were compared using the Kaplan-Meier method with log-rank test and post hoc pairwise comparison. The 95% confidence intervals (CIs) were estimated using the means and standard errors. The Cox proportional hazards model was used for multivariate analysis. The p-values of <0.05 were considered significant. For all statistical analyses, we used IBM SPSS ver. 23.0 (IBM Corp., Armonk, NY, USA).
Go to :
Results
1. Baseline characteristics of patients with HLH
During the study period, 60 patients (35 males and 25 females) fulfilled the criteria for HLH. The median age at diagnosis of HLH was 7.0 years (range, 0.1–83 years). In the study population, 35 patients were children (aged <18 years) and 25 patients were adults (aged ≥18 years). Although HLH occurred in all age groups, more than 50% of the patients (34/60, 56.7%) developed HLH under 10 years of age (Fig. 1A). The median follow-up duration was 8.5 months (range, 0–204 months). On dividing the patients according to age, it was observed that the number of adult patients with HLH increased each year from 2001 to 2019 (Fig. 1B).
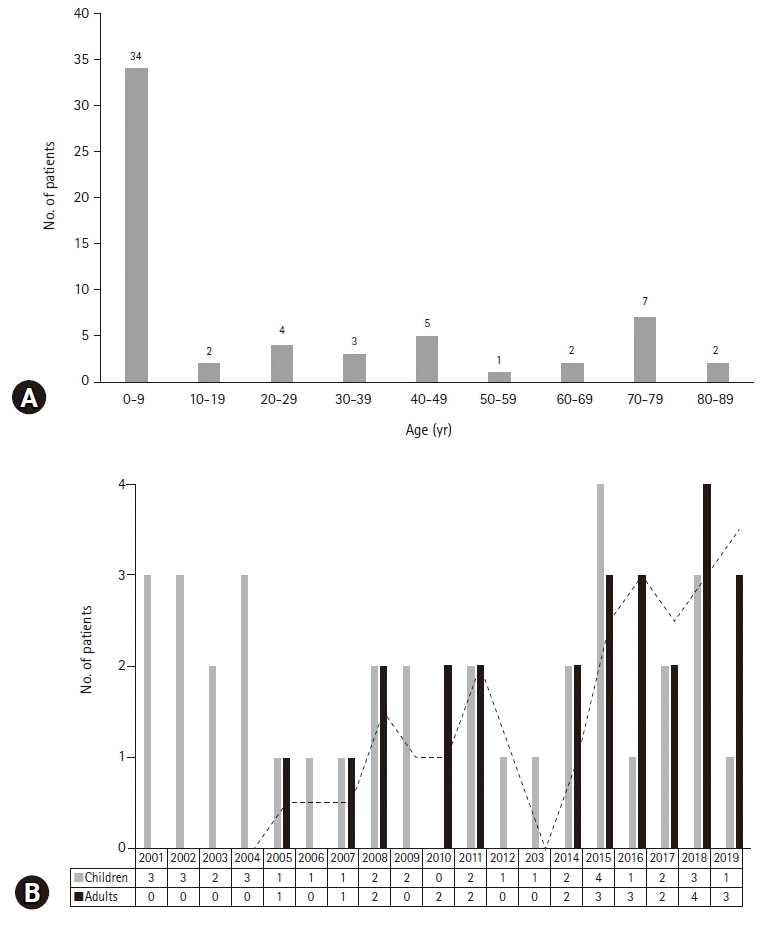
The baseline characteristics of the HLH patients are presented in Table 1. Among 60 patients, 15 underwent genetic testing for HLH, four of whom were confirmed as having primary HLH. Out of the four patients with primary HLH, three patients had FLH while one had XLP1. Forty-eight patients had secondary HLH; 20 patients with infection-associated HLH, 18 with neoplasm-associated HLH, and 10 with autoimmune-associated HLH. In the remaining eight patients, the cause of HLH was unknown. In secondary HLH, EBV was the most common cause of infection-associated HLH (6/20, 30.0%), lymphoma was the most common of the neoplasm-associated HLH (10/18, 55.6%), and Kawasaki disease was the most common cause of autoimmune-associated HLH (5/10, 50.0%). With respect to the treatment regimen, 28 patients were treated with HLH-2004- or HLH-94-based immunochemotherapy, and the remaining 32 patients were treated with other regimens; corticosteroid was the commonly used medication (20/32, 62.5%).
Table 1.
Baseline characteristics of patients with HLH
![]()
Each of the 60 HLH patients in this study underwent BM aspiration and biopsy. None of them underwent biopsy of organs (lymph node, spleen, or liver) other than BM. Among 60 patients, hemophagocytosis was found in the BM of 56 patients, but not in that of four patients. There were 26 patients who were included in the BM study, despite not meeting the five diagnostic criteria for HLH; the median hemoglobin level in these patients was 87 g/L (range, 51–157 g/L), the median neutrophil count was 1.95×109/L (range, 0.18–14.09×109/L), and the median platelet count was 70×109/L (range, 15–108×109/L). The blood test results of the remaining 34 patients were as follows: the median hemoglobin level was 96 g/L (range, 65–142 g/L), the median neutrophil count was 0.85×109/L (range, 0.06–7.71×109/L), and the median platelet count was 59×109/L (range, 23–384×109/L). There were no statistically significant differences in hemoglobin, neutrophil count, and platelet count between the two groups (p=0.200, p=0.051, and p=0.617, respectively).
2. Comparison of the causes of HLH according to the age
The underlying pathophysiology of HLH according to the age group is shown in Fig. 2. In children with HLH, infection was the most common cause (14/35, 40.0%), followed by autoimmune diseases (6/35, 17.1%), uncertain causes (7/35, 20.0%), neoplasm (5/35, 14.3%), and primary HLH (3/35, 8.6%). In adults with HLH, neoplasm was the most common cause (13/25, 52.0%), followed by infection (6/25, 24.0%), autoimmune diseases (4/25, 16.0%), and primary HLH or uncertain cause (both, 1/25, 4.0%). A higher proportion of children had infection as the cause of HLH (40.0%) than that of adults (24.0%) (p=0.029). Moreover, neoplasm-associated HLH was more common in adults (52.0%) than in children (14.3%) (p=0.004). Conversely, the difference in the prevalence of autoimmune diseases, uncertain causes, and primary HLH was not significant between children and adults (p=1.000, p=0.123, and p=0.634, respectively).
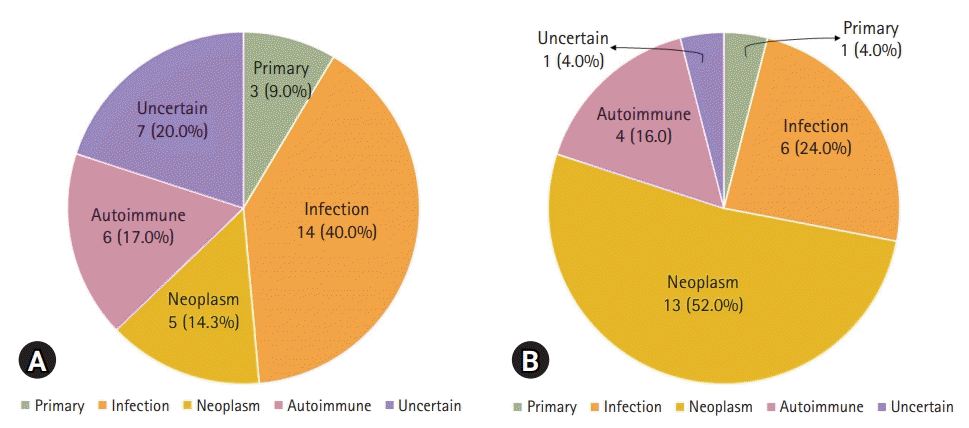
3. Comparison of the variables between pediatric and adult patients with HLH
A comparison of the variables of clinical manifestations, laboratory tests, and treatment regimens between pediatric and adult patients with HLH is presented in Table 2. In the case of clinical manifestations, the prevalence of splenomegaly was higher in adults (24/25, 96.0%) than in children (24/35, 68.6%) (p=0.010). Conversely, the prevalence of hepatomegaly was higher in children (28/35, 80.0%) than in adults (10/25, 40.0%) (p=0.003). The levels of the liver enzyme alanine aminotransferase were also higher in children (median, 232 U/L; range, 14–1,230 U/L) than in adults (median, 120 U/L; range, 8–2,019 U/L) (p=0.024). With respect to the treatment regimen, the proportion of patients treated with HLH-2004/94 protocol was higher among children (28/35, 80.0%) than that in adults (0/25, 0%) (p<0.001). Differences in the variables for the rest of the clinical features and test findings between children and adults were not significant.
Table 2.
Comparison of the variables between children and adults with HLH
| Variable | Child (n=35) | Adult (n=25) | p-value |
|---|---|---|---|
| Sex (male:female) | 21:14 | 14:11 | 0.481 |
| EBV | 7/34 (20.6) | 4/20 (20.0) | >0.050 |
| Fever | 35 (100) | 25 (100) | - |
| Splenomegaly | 24 (68.6) | 24 (96.0) | 0.010 |
| Hepatomegaly | 28 (80.0) | 10 (40.0) | 0.003 |
| Neurologic symptom or sign | 4 (11.4) | 4 (16.0) | 0.708 |
| Rash | 13 (37.1) | 5 (20.0) | 0.253 |
| Hemoglobin (g/L) | 90 (51–121) | 89 (64–157) | 0.685 |
| Neutrophil count (×109/L) | 0.97 (0.06–14.1) | 1.00 (0.18–10.2) | 0.589 |
| Platelet count (×109/L) | 67 (25–384) | 52 (15–158) | 0.184 |
| Triglyceride (mg/dL) | 219 (47–742) | 244 (128–274)a) | 0.988 |
| Fibrinogen (g/L) | 1.32 (0.70–4.22)b) | 2.18 (0.45–6.18) | 0.051 |
| Ferritin (μg/L) | 3,225 (185–204,887) | 2,450 (764–19,640)c) | 0.765 |
| sCD25 (U/mL) | 4,745 (825–27,060)d) | 2,817e) | 0.714 |
| Hemophagocytosis | 32 (91.4) | 24 (96.0) | 0.634 |
| AST (U/L) | 450 (22–5,137) | 221 (11–2,237) | 0.063 |
| ALT (U/L) | 232 (14–1,230) | 120 (8–2,019) | 0.024 |
| LDH (U/L) | 1,565 (266–5,861)f) | 1,660 (262–6,369)c) | 0.666 |
| Total bilirubin (mg/dL) | 1.2 (0.1–11.8) | 1.1 (0.3–8.2) | 0.584 |
| Direct bilirubin (mg/dL) | 0.6 (0.0–8.8) | 1.3 (0.2–6.2)g) | 0.617 |
| PT (sec) | 12.9 (11.7–31.6)b) | 13.7 (11.0–19.4) | 0.758 |
| INR | 1.19 (1.00–2.85)b) | 1.18 (0.93–1.85) | 0.515 |
| aPTT (sec) | 40.1 (25.5–200.0)b) | 39.3 (21.6–68.8) | 0.921 |
| HLH-2004/94 treatment | 28 (80.0) | 0 | <0.001 |
Values are presented as number only, number (%), or median (range).
HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein–Barr virus; sCD25, soluble CD25; AST, aspartate transaminase; ALT, alanine transaminase; LDH, lactate dehydrogenase; PT, prothrombin time; INR, international normalized ratio; aPTT, activated partial thromboplastin time.
![]()
4. Comparison of the variables between survival and death groups of patients with HLH
A comparison of the variables between survival and death groups is presented in Table 3. The median age at diagnosis was lower in the survival group (median, 5.3 years; range, 0.1–76.5 years) than that in the death group (median, 39.3 years; range, 0.1–89 years) (p=0.007). The prevalence of neoplasm-induced HLHs in the survival group (5/37, 13.5%) was lower than that in the death group (13/23, 56.5%) (p=0.001). The hemoglobin levels were higher in the survival group (median, 99 g/L; range, 51–157 g/L) than in the death group (median, 86 g/L; range, 64–116 g/L) (p=0.006). The platelet levels were higher in the survival group (median, 67×109/L; range, 25–384×109/L) than in the death group (median, 51×109/L; range, 15–117×109/L) (p=0.030).
Table 3.
Comparison of the variables between the survival group and death group
| Variable | Survival group (n=37) | Death group (n=23) | p-value |
|---|---|---|---|
| Sex (male:female) | 20:17 | 15:8 | 0.432 |
| Age (yr) | 5.3 (0.1–76.5) | 39.3 (0.1–89) | 0.007 |
| Primary HLH | 1 (2.7) | 3 (13.0) | 0.153 |
| Infection-associated HLH | 17 (45.9) | 3 (13.0) | 0.011 |
| Neoplasm-associated HLH | 5 (13.5) | 13 (56.5) | 0.001 |
| Autoimmune-associated HLH | 9 (24.3) | 1 (4.3) | 0.073 |
| Uncertain cause HLH | 5 (13.5) | 3 (13.0) | >0.050 |
| EBV | 6/32 (18.8) | 5/22 (22.7) | 0.743 |
| Fever | 37 (100) | 23 (100) | - |
| Splenomegaly | 27 (73.0) | 21 (91.3) | 0.107 |
| Hepatomegaly | 25 (67.6) | 13 (56.5) | 0.421 |
| Neurologic symptom or sign | 4 (10.8) | 4 (17.4) | 0.468 |
| Rash | 13 (35.1) | 5 (21.7) | 0.387 |
| Hemoglobin (g/L) | 99 (51–157) | 86 (64–116) | 0.006 |
| Neutrophil count (×109/L) | 0.9 (0.13–14.1) | 1 (0.06–10.2) | 0.638 |
| Platelet count (×109/L) | 67 (25–384) | 51 (15–117) | 0.030 |
| Triglyceride (mg/dL) | 228 (47–742) | 206 (48–347) | 0.335 |
| Fibrinogen (g/L) | 1.48 (0.70–5.00)a) | 1.40 (0.45–6.18) | 0.893 |
| Ferritin (μg/L) | 3,200 (528–204,887) | 2,935 (185–57,186) | 0.850 |
| sCD25 (U/mL) | 4,285 (825–10,013)b) | 17,647 (1,139–27,060)c) | 0.142 |
| Hemophagocytosis | 33 (89.2) | 23 (100) | 0.288 |
| AST (U/L) | 307 (11–5,137) | 285 (22–4,829) | 0.238 |
| ALT (U/L) | 178 (8–2,019) | 120 (14–1,043) | 0.110 |
| LDH (U/L) | 1,749 (262–5,861)d) | 1,164 (437–15,062)e) | 0.110 |
| Total bilirubin (mg/dL) | 1.0 (0.3–11.8) | 1.2 (0.1–8.2) | 0.171 |
| Direct bilirubin (mg/dL) | 0.6 (0.1–8.8) | 1.4 (0–6.2)f) | 0.710 |
| PT (sec) | 12.9 (11.0–22.3)a) | 13.8 (12.9–16.5) | 0.229 |
| INR | 1.17 (0.99–2.07)a) | 1.27 (0.93–2.85) | 0.368 |
| aPTT (sec) | 40.1 (26.0–79.6)a) | 38.6 (21.6–200.0) | 0.681 |
| HLH-2004/94 treatment | 21 (56.8) | 7 (30.4) | 0.064 |
Values are presented as number only, number (%), or median (range).
HLH, hemophagocytic lymphohistiocytosis; EBV, Epstein–Barr virus; sCD25, soluble CD25; AST, aspartate transaminase; ALT, alanine transaminase; LDH, lactate dehydrogenase; PT, prothrombin time; INR, international normalized ratio; aPTT, activated partial thromboplastin time.
![]()
5. Survival of HLH patients
The 5-year OS rate of all patients with HLH (n=60) was 59.9% (95% CI, 46.6–73.2) (Fig. 3A). The comparison of 5-year OS rates according to the variables by univariate analysis is shown in Table 4. According to age, the difference in 5-year OS between children (72.1%; 95% CI, 56.4–87.8) and adults (37.4%; 95% CI, 11.3–63.5) was significant (p=0.007). Based on the HLH classification, the 5-year OS rates in patients with primary, infection-associated, neoplasm-associated, autoimmune-associated, and uncertain cause of HLH were 25.0% (95% CI, 0–67.5), 85.0% (95% CI, 69.3–100), 26.7% (95% CI, 3.6–49.8), 87.5% (95% CI, 64.6–100), and 62.5% (95% CI, 29–96), respectively. Comparatively, the difference in the 5-year OS between the primary and infection-associated HLHs was significant (p=0.024). The difference in the 5-year OS between the neoplasm-associated and infection-associated HLHs was significant (p=0.001). Additionally, the difference in the 5-year OS between the neoplasm-associated and autoimmune-associated HLHs was significant (p=0.010). The 5-year OS rate, according to the HLH classification, is also shown as a graph in Fig. 3B. As per laboratory results, hemoglobin level of <90 g/L or platelet count of <50×109/L at initial diagnosis of HLH were risk factors for a low 5-year OS rate.
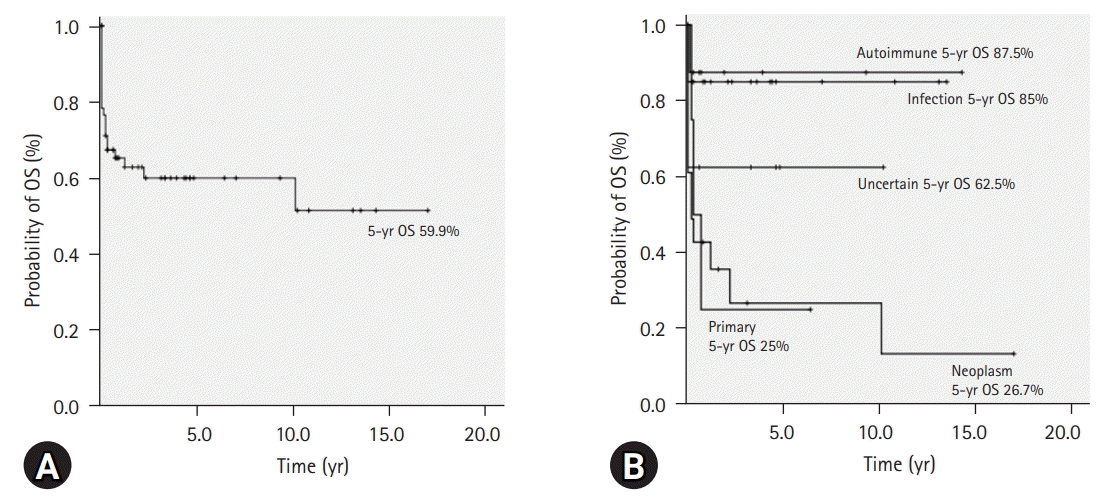 | Fig. 3.(A) In all patients with hemophagocytic lymphohistiocytosis (HLH) (N=60), the 5-year overall survival (OS) rate is 59.9% (95% CI, 46.6–73.2). (B) The 5-year OS according to the classification of HLH.
|
Table 4.
Comparison of OS rate of patients with HLH according to the variables by univariate analysis
![]()
In terms of the treatment regimen, the 5-year OS rate in patients treated with HLH-2004 protocol was 74.5% (95% CI, 54.9–94.1), in those treated with HLH-94 protocol was 68.6% (95% CI, 32.1–100), and in patients who received other treatments was 45.8% (95% CI, 25.0–66.6). The difference in the 5-year OS rate between HLH-2004 and HLH-94 protocol groups was not significant (p=0.904). The difference in the 5-year OS rate between patients treated with HLH-2004/94 (73.1%; 95% CI, 55.9–90.3) and other treatments (45.8%; 95% CI, 25.0–66.6) was significant (p=0.017). By multivariate analysis using the Cox proportional hazards model, neoplasm-induced HLH (hazard ratio, 4.446; 95% CI, 1.876–10.538; p=0.001) and platelet count of <50×109/L at initial diagnosis of HLH (hazard ratio, 3.298; 95% CI, 1.373–7.92; p=0.008) were identified as independent risk factors of death in patients with HLH.
Go to :
Discussion
In this study, we retrospectively investigated the causes and survival rates in children and adults with different HLH subtypes. Moreover, we analyzed the differences in clinical characteristics between children and adults with HLH. Based on the statistical analyses of this study, HLH was observed primarily in children (median age, 7 years); however, during the period from 2001 to 2019, the number of adult patients diagnosed with HLH had increased. During the review of the medical data for this retrospective study, several patients suspected to have HLH and treated with corticosteroids or immunoglobulins were not included in this study because they had not undergone all tests corresponding to the diagnostic criteria of HLH. Therefore, the increase in the number of diagnoses of adult patients with HLH during the study period is not exactly an increase in HLH cases, but the result of active examinations with increasing awareness among the medical staff. The awareness of the symptoms of HLH as well as the diagnostic criteria is crucial for physicians because early diagnosis and prompt therapy are crucial for the outcome given the life-threatening nature of HLH.
As with the previous Korean nationwide pediatric HLH data from the Korea Histiocytosis Working Party [13], EBV was the main cause of secondary HLH. This finding is an example of the higher rates of EBV-induced secondary HLH in the Asian population [19]. Apart from EBV, various other organisms may also cause HLH, as reported by previous studies [13,16]. In this study, infection was the main cause of HLH in children. However, neoplasm (mainly lymphoma) was the main cause of HLH in adults, which is consistent with other international data [16,19]. In terms of clinical manifestations and laboratory tests, hepatomegaly with increased aminotransferase was predominant in pediatric patients with HLH; conversely, splenomegaly was more apparent in adult patients with HLH in this study. This clinical aspect is also considered to be related to the difference in causative agents between children and adults.
In the case of pediatric HLH patients in this study, the 5-year OS rate was 72.1% (n=35). In a previous nationwide data of pediatric HLH in Korea, the 5-year OS rate was 68% [13]. Although it is difficult to directly compare these results because of the small number of subjects in this study, it can be said that the survival rates were relatively similar. The difference in the 5-year OS between the HLH-2004 and HLH-94 protocols was not significant (p=0.904). These data are the same as previously published data [11,12]. However, we can consider the improvement of supportive and conservative therapy over time from HLH-94 to HLH-2004. In the case of adult HLH patients, specific therapeutic guidelines have not yet been confirmed worldwide. It seems that each institution decides the treatment direction according to the patient’s condition [16]. As in our study, most adult HLH patients were treated with corticosteroids, immunoglobulin G, or cyclosporin A [16]. Thus, there was no remarkable difference between the previous data and the present study. Recently, recommendations for adult HLH patients have been published that can be useful in determining treatment directions [20].
Although an accurate comparison is difficult considering the very small number of primary HLHs in this study, the most common cause of primary HLH was FHL type 3 with UNC13D variants, similar to previous Korean data [13]. Interestingly, one patient with FHL type 2 (PRF1 variant) in this study was a 28.3-year-old man. Initially, this patient was diagnosed with T-cell lymphoma that progressed aggressively, and he met the criteria of HLH with multiple organ failure. Because of this extraordinary disease course, the physician performed a genetic test and he was eventually diagnosed with adult FHL. Although primary HLH is commonly diagnosed in children, several examples of genetically confirmed HLH in adults have recently been reported [21-23]. Allogenic stem cell transplantation is a recommended therapy for adult primary HLH [23]. Furthermore, a 9.2-year-old pediatric patient with XLP1 (SH2D1A variant) was initially diagnosed with EBV-associated lymphoproliferative disease, which also demonstrated very aggressive features of HLH with multiple organ failure. As there was no pathogenic variant based on the conventional genetic tests for PRF1 and UNC13D, the pediatrician ordered next-generation sequencing, and finally, he was diagnosed with XLP1. These two patients with FHL would have been classified as neoplasm-associated secondary HLH unless additional genetic testing was performed. Therefore, physicians must not only perform all tests in the diagnostic criteria but also consider genetic testing for the accurate diagnosis of HLH. In this study, some patients with FHL may have been classified as patients with neoplasm-associated HLH or uncertain cause HLH because genetic testing had not been thoroughly conducted. Additionally, patients with uncertain cause HLH may include those who had not been thoroughly screened for underlying infective organisms.
In all patients with HLH, the 5-year OS rate was 59.9% in this study. Univariate analysis showed that the OS rate of HLH patients was affected by the age of diagnosis, type of HLH, treatment with HLH protocol, anemia, or thrombocytopenia. However, the underlying cause, neoplasm-associated HLH, was the only independent risk factor for death by multivariate analysis in this study. Therefore, optimal management for patients with neoplasm-associated HLH requires discussion. Treatment algorithms for HLH, such as HLH-94 and HLH-2004, are mainly based on pediatric protocols, which may result in unnecessary toxicity or overtreatment in adults [20]. Thus, an age-dependent modified therapeutic approach, such as individualized tailoring or reduction of treatment duration, must be considered [20].
This study has some limitations. First, this study was performed retrospectively; thus, there is a possibility of selection bias among the study populations. During the review of the chart, several children as well as adults had not undergone tests such as soluble CD25 or NK cell activity. Although several patients had the characteristics of HLH, they were not enrolled in this study because they did not undergo the tests and did not meet the diagnostic criteria. Therefore, HLH may have been underdiagnosed during the study period. Second, adult patients with HLH were enrolled according to the HLH-2004 criteria, which focused on pediatric patients to clarify the scope of the study subjects. The HLH-2004 criteria have not yet been formally validated for adults, and thus, it continues to be based on expert opinion [20]. Various case series have used modified HLH-2004 criteria [24].
In conclusion, this is the first Korean study that analyzed the underlying causes and patients’ survival in both children and adults with HLH. Additionally, we compared the clinical characteristics according to the age group and analyzed the survival rate according to the HLH subtypes. The most common cause of HLH in children was infection, while that in adults was neoplasm. The 5-year OS rate for all HLH patients was 59.9%. Neoplasm-induced HLH and platelet count of <50×109/L at the time of diagnosis were independent risk factors for poor prognosis in patients with HLH. Therefore, it is essential to accurately identify the cause in patients with HLH. In addition, the optimal treatment regimen for neoplasm-induced HLH requires further discussion to improve the outcome for adult patients.
Go to :
 XML Download
XML Download