

 PDF
PDF Citation
Citation Print
Print



Introduction
Oculocutaneous albinism (OCA) is a group of rare genetically heterogeneous disorders. OCA is an autosomal recessive disorder associated with mutations in genes which control the biosynthesis of the melanin pigment [1]. The pigmentary system is dependent on the production of melanin, the light-absorbing biopolymer found within ocular, epidermal, and follicular melanocytes [2]. OCA is characterized by hypopigmentation of the eyes, skin and hair, which results in ocular abnormalities and a risk of developing skin cancer. All patients with OCA are accompanied by ocular abnormalities, such as nystagmus, reduced visual acuity, photophobia, strabismus, foveal hypoplasia, hypopigmentation of the iris, and color vision impairment [3]. Currently, there is no ophthalmologic procedure or drug that prevents the clinical features of OCA. The clinical spectrum of OCA varies, ranging from mild hypomelanosis to complete lack of melanin production, which is mainly associated with tyrosine metabolism [4].
To-date, seven types of OCA (OCA1-7) have been identified, depending on a different genetic defect. OCA2, which is the most common type of albinism worldwide [5], is caused by pathogenic variants of OCA2 gene [6]. The OCA2 locus maps to chromosome 15q and is the human homolog of the mouse pink-eyed dilution gene encoding the P protein [7-9]. The mutation types causing OCA2 are variable, including missense/nonsense single nucleotide variations (SNVs), splicing variations, small indels, and large indels. So far, 184 relevant mutations have been reported in Human Gene Mutation Database (HGMD, professional 2019.4). The most common OCA2 mutation is a 2.7-kb deletion on exon 7, which is found in many affected individuals of sub-Saharan African descent [10]. Other OCA2 mutations, including missense/nonsense SNVs and small deletions, are more common in other populations (https://ghr.nlm.nih.gov/gene/OCA2).
Prevalence of OCA2 is approximately 1:38,000 to 1:40,000 in most populations throughout the world. The exception to this is the African population, in which the prevalence is estimated at 1:1,500 to 1:3,900 [10,11]. Here we report a new type of OCA in two unrelated Korean families with the same OCA2 mutation.
Go to : 
Case
Participants were sourced from two, unrelated Korean families comprising four generations. This study was approved by the Institutional Review Board of the Kyungpook National University Hospital (IRB No: KNUH 2016-06-011). Written informed consents were obtained from the patient’s parents for publication of this case report and accompanying images.
A 4-year-old Korean male patient came to seek medical attention for genetic diagnosis of albinism. The patient had ivory skin, medium blond hair, and light brown eyes (Fig. 1A). The father of the patient was also diagnosed with OCA when he was young (Fig. 1B). On ophthalmologic examination, best-corrected visual acuity (BCVA) was 20/50 for both eyes. He presented with esotropia and reduced stereoscopic vision. The iris was hypopigmented, but iris translucency was not evident on slit lamp examination. Fundus examination showed hypopigmented fundi (Fig. 1C). The patient was born with pale white skin and blond hair, both of which had acquired increased pigmentation with age. The color of his eyes, skin and hair gradually became darker and attained normal pigmentation in his late twenties. At the time of the visit, the father showed normal black hair and tan skin (Fig. 1A). The albinism showed an autosomal dominant inheritance pattern through the history of the family: the father, one of his aunts, grandfather, and great-grand mother (Fig. 1D).
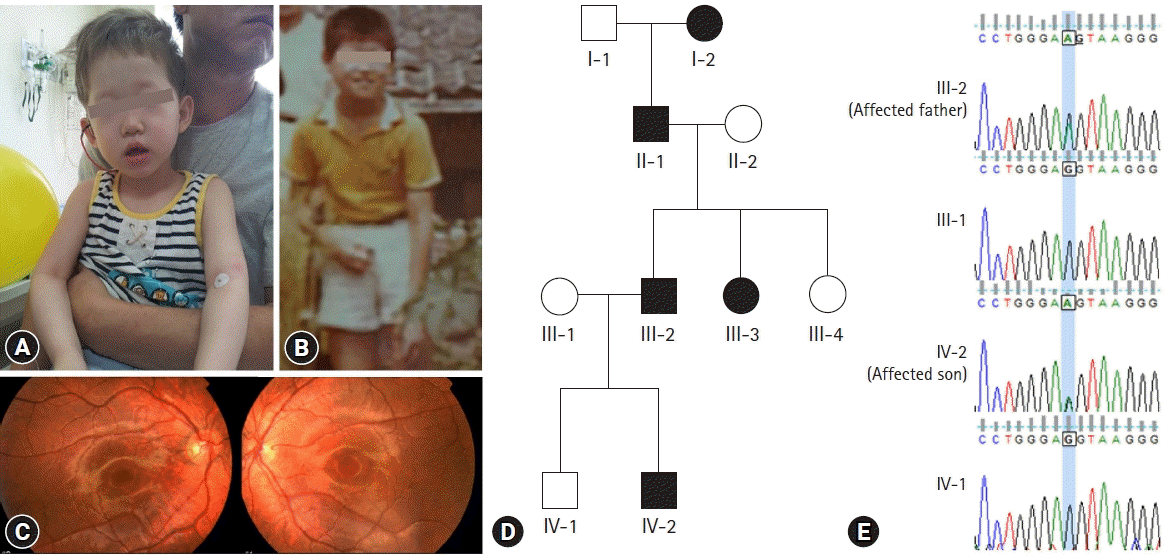 | Fig. 1.(A) General appearance of the affected son and the affected father of the first family at the time of visit. (B) The father of the patient also had oculocutaneous albinism when he was young. (C) Hypopigmented fundi of the affected son. (D) The pedigree shows autosomal dominant inheritance. (E) Chromatograms of family members showing the same OCA2 mutation in the affected individuals.
|
A 6-year-old Korean female patient was referred for evaluation of hypopigmentation. She had ivory skin, dark blond hair, and light brown eyes (Fig. 2A). Her younger sister had normal pigmentation (Fig. 2B). She had pale white skin and blond hair when she was born (Fig. 2C). On ophthalmologic examination, BCVA was 20/25 for both eyes and fundus examination showed hypopigmented fundi (Fig. 2D). A gradual increase in pigmentation was observed in the color of her eyes, skin and hair compared to the time of her birth. Similar to the father of the male patient described earlier, her father had OCA when he was young, the characteristics of which became darker and attained normal pigmentation in his late twenties. The albinism showed an autosomal dominant inheritance pattern through the father, grandmother, great-granduncle, and great-grandmother (Fig. 2E). The affected individuals had similar characteristics which displayed progressive darkening and completely normal pigmentation by their late twenties.
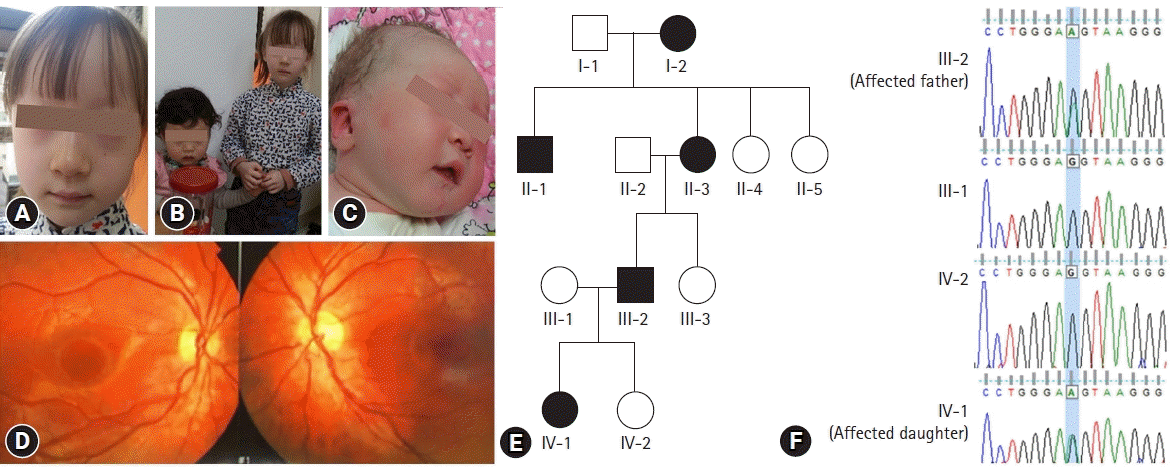 | Fig. 2.(A) General appearance of the affected daughter at the time of visit. (B) General appearance of the affected daughter and her unaffected sister. (C) The affected daughter had pale white skin and blond hair when she was just born. (D) Hypopigmented fundi of the affected daughter. (E) The pedigree shows autosomal dominant inheritance. (F) Chromatograms of family members showing the same OCA2 mutation in the affected individuals.
|
Whole exome sequencing and in-silico analysis revealed a novel mutation, OCA2 c.2338G>A p.(G780S) (NM_000275). In-silico analysis was performed using bioinformatics software programs such as SIFT (http://sift.jcvi.org/), polyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and phastCons score. Prediction results were deleterious (0.00) in SIFT and damaging (1.00) in polyPhen-2, indicating that the amino-acid substitutions have a high probability of pathogenicity. G780S was extremely conserved in multiple species with phastCons scores of 1.0, and has never been reported in the HGMD, HGMD Professional database, dbSNP, or the 1000 Genomes database with a minor allele frequency of 0. Sanger sequencing of p.G780S validated the same mutation in the affected individuals which was not identified in the family members with normal phenotype (Fig. 1E, 2F).
Go to :
Discussion
OCA2 encodes a pigment cell-specific, 12-transmembrane domain protein with homology to ion permeases. G780S is located in one of the extracellular topological domains of OCA2. Several studies demonstrated the function of OCA2 as a positive regulator of pH neutralization and melanogenesis promotion [12,13]. TYR is the key enzyme for melanogenesis which is solely dependent on neutral pH. Ion transport proteins such as OCA2, SLC45A2, and TPC2 function together to promote TYR function in a so called “combinational function of ion transport proteins” for the maintenance of pH [14]. OCA2 induces large outward Cl− conductance which is essential for maintaining neutral pH for melanosomes [14]. SLC45A2 and TPC2 act as modifier genes of OCA2. Modifier genes are defined as genes that affect the phenotypic properties of other genes. Genetic modifiers can affect penetrance, dominance, expressivity, and pleiotropy. The P protein is not composed of dimers and seems unlikely to have a dominant-negative effect. It might represent a gain-of-function due to other historical genetic factors of melanogenesis in these families.
Known pathogenic mutations of OCA2 include V443I, P743L, and A481T. Patients with compound heterozygous mutations of V443I and P743L had severe OCA2 phenotypes [15]. OCA2 A481T has previously been reported to result in partial function of the P protein [15-17]. Melanin production gradually increased in patients with OCA2 A481T during childhood, but no case had been reported where a patient attained normal pigmentation. The patients with OCA2 A481T had a compound heterozygous pathogenic OCA2 mutation in the other allele, and heterozygous carriers of OCA2 A481T did not show hypopigmentation [18]. Most carriers are normally asymptomatic as pathologic mutation in one copy of the OCA2 gene does not result in OCA. However, some individuals have been reported to have mild phenotypes, such as a small degree of iris transillumination, or hair and skin hypopigmentation in a heterozygous state [18]. In another study, carriers of N476D of OCA2 presented with a mild hypopigmentation phenotype, which was particularly obvious at a young age [19].
Affected individuals in this study are different from those previously reported in two aspects: an inheritance pattern and clinical presentation. All reported patients with OCA to-date have shown an autosomal recessive inheritance pattern, while our patients showed an autosomal dominant inheritance pattern. There has never been a reported case of a patient who attained normal pigmentation, while our patients displayed completely normal pigmentation in their late twenties. Individuals with OCA2 mutation can acquire small amounts of pigment with age, but this does not vary substantially from adolescence to adulthood [20].
In summary, we report a novel OCA2 gene mutation found in two OCA families. We hypothesize that OCA2 G780S not only acts as a pathogenic variant of OCA, but also induces pigmentation by enhancing melanogenesis gene expression of other modifier genes, such as SLC45A2 and TPC2. These findings may provide further understanding of melanin biosynthesis and new treatments of OCA.
Go to :
 XML Download
XML Download