

 PDF
PDF Citation
Citation Print
Print



Introduction
Monogenic diabetes is caused by mutations in one of the genes that control insulin levels [1,2]. Forms of monogenic diabetes include neonatal diabetes, maternally inherited diabetes with deafness and genetic syndromes, such as Bardet-Biedl syndrome, and Wolfram syndrome; however, maturity-onset diabetes of the young (MODY) and neonatal diabetes are the most common. MODY was first described in 1974 by Tattersall [3] as a mild familial diabetes with dominant mode of inheritance. MODY is a clinically heterogeneous group of monogenic diabetes mellitus characterized by ß-cell dysfunction. The diagnostic criteria reported in 2008 include onset before 25 years of age in at least one of the family members, ß-cell dysfunction without autoantibodies, and family history of autosomal dominant diabetes for at least two generations [4]. In the 1990s, molecular methods for the diagnosis of MODY were introduced, after which the mutations associated with the disease were identified. To date, MODY-associated mutations have been reported in 14 different genes (Table 1) [5-10]. MODY is the most common type of monogenic diabetes and comprises between 2% to 5% of all diabetes cases in Europe [11,12]. Among these genes, mutations in HNF1A, GCK, HNF4A, and HNF1B are the underlying cause in more than 95% cases of MODY; the other mutations are rare in the Caucasian population [12-14]. There is a discrepancy in the disease-causing locus between the Caucasian and Asian patients with MODY. Moreover, the exact prevalence of the disease in the Asian population has not been reported. This review summarizes the current understanding of MODY and discusses the Asian studies of the disease.
Table 1.
Genes related to MODY and clinical characteristics of each MODY subtype
![]()
Go to : 
MODY2 (GCK-MODY)
Glucokinase (GCK) is a major enzyme in glucose metabolism, which catalyzes the conversion of glucose to glucose-6 phosphate, and thus, controls glucose-mediated insulin secretion. Until 2009, more than 600 mutations in GCK had been identified in over 1,400 families [15]. The inactivating heterozygous mutations in GCK elevate the glucose threshold for insulin secretion resulting in mild fasting hyperglycemia (5.6–8.0 mmol/L, glycosylated hemoglobin range of 5.6%–7.3%) [16]. Mutations in GCK are some of the most common causes of MODY, and affect 32% of the total number of patients with MODY in the United Kingdom [17]. Patients with GCK-MODY are usually asymptomatic; hence, the majority are diagnosed through routine examination, such as urine glucose screening at school or during pregnancy. Mutations in GCK were found in 40%–50% of incidental hyperglycemia cases in children; therefore, the prevalence of GCK-MODY is high in countries where the school glucose screening test is performed [18,19]. However, only 2.5% of 40 studies of MODY and early onset type 2 diabetes in Korea reported GCK mutations [20]. This implies that there might be a discrepancy between the Caucasian and Asian patients. The clinical manifestations of GCK-MODY may be mild and non-progressive as long-term complications rarely develop despite chronic mild hyperglycemia. Therefore, patients with this mutation usually do not require treatment, except during pregnancy [21].
Go to :
MODY3 (HNF1A-MODY)
Hepatocyte nuclear factor 1α (HNF1A) is a transcription factor expressed in various organs, such as the pancreas, kidney, liver, and intestine. HNF1A knockout mice develop diabetes due to impaired glucose-induced insulin secretion [22,23]. Prevalence of this mutation is the highest in Europe, North America, and Asia [17,20,24]. Over 400 different HNF1A mutations have been identified in approximately 1,200 families; among these, a mutation in exon 4 of the gene (P291fsinsC) is the most frequently observed [25,26]. These mutations alter the expression of proteins related to glucose transport, such as glucose transporters, as well as that of key enzymes involved in mitochondrial glucose metabolism. In HNF1A-knockout mice, reduced ß-cell proliferation and increased apoptosis leads to a progressive decline in ß-cell function [27]. HNF1A mutations have high penetrance, with almost 63% of their carriers developing diabetes by the age of 25, and almost 96% by the age of 55 [28]. Since HNF1A is also expressed in tissues other than the pancreas, patients with HNF1A-MODY can display extra-pancreatic manifestations such as glycosuria, which can develop even before the onset of diabetes because of a low renal threshold for glucose [29]. Hyperglycemia induced by heterozygous HNF1A mutations might be deteriorating and progressive, and the risk of developing long-term complications in HNF1A-MODY is similar to that in type 1 and type 2 diabetes [30]. Therefore, rigorous glucose control is needed in these patients.
Patients with HNF1A-MODY show marked sensitivity to the oral hypoglycemic agent sulfonylurea, which elicits a five-fold greater response than metformin although the two agents show similar efficacy in type 2 diabetes [31]. Consequently, many patients with HNF1A-MODY achieve better glycemic control with sulfonylurea than with insulin treatment [32,33]. Therefore, low-dose sulfonylurea should be considered the first-line treatment for HNF1A-MODY, although some patients might need additional insulin therapy as the diabetes progresses [34].
Go to :
MODY1 (HNF4A-MODY)
HNF4A is a transcription factor primarily expressed in the liver and, to a lesser extent, in the kidney and pancreas. HNF4A regulates the transcription of genes involved in glucose transport and metabolism [35]. Mutations in the HNF4A gene are uncommon and account for only approximately 3%–5% of all MODY cases; more than 100 HNF4A mutations have been identified in 173 families [25,36]. Patients with heterozygous HNF4A mutations display progressive ß-cell dysfunction similar to that observed in patients with HNF1A mutations. Fetal heterozygous HNF4A mutation results in diazoxide-responsive form of neonatal hyperinsulinemic hypoglycemia and subsequent macrosomia [31]. Therefore, close monitoring of the baby of an affected mother is recommended. The hyperinsulinemia usually resolves during infancy and the insulin production gradually decreases leading to the development of diabetes in adolescence [24]. Unlike HNF1A-MODY, HNF4A-MODY is not associated with glycosuria. Instead, low levels of apolipoproteins (apoAII, apoCIII, and apoB) can be a clue to diagnosing this subtype [37]. HNF4A-MODY is characterized by sensitivity to sulfonylureas similar to that of HNF1A-MODY; therefore, low-dose sulfonylurea is recommended as the first-line treatment [31].
Go to :
MODY5 (HNF1B-MODY)
HNF1B is a transcription factor associated with early organogenesis of the pancreas, kidney, liver, lungs, gut, and genito-urinary tract [38]. Patients with HNF1B mutations develop abnormalities in all these organs; however, renal manifestations, such as renal cysts, renal dysplasia, renal tract malformations, and familial hypoplastic glomerulocystic kidney disease, are the most common [39,40]. The association of renal cysts and diabetes mellitus with mutations in the HNF1B gene is termed the renal cysts and diabetes syndrome. Renal dysfunction is usually developed by the age of 45, and approximately 50% of the patients progress to end-stage renal failure requiring renal replacement therapy without diabetic renal disease [41]. Therefore, carriers of HNF1B mutations should be monitored for the development of diabetes and non-diabetic nephropathy. Diabetes associated with MODY5 develops in adolescence or early adulthood and presents with hepatic insulin resistance before progressing to insulin-dependent status due to pancreatic hypoplasia. HNF1B mutations can reduce the birth weight by up to 900 g [42,43]. In contrast to patients with MODY3, those with MODY5 progress to insulin-dependent status and do not respond to sulfonylurea; therefore, they usually require early insulin therapy. Patients with HNF1B mutations manifest highly variable phenotypes, which might even differ between family members carrying the same mutation. Hence, patients with HNF1B-MODY should seek endocrinology, as well as nephrology, urology, and gynecology consultation.
Go to :
MODY4 (IPF1-MODY)
Insulin promoter factor 1 (IPF1) is a transcription factor that regulates ß-cell development and insulin expression in pancreatic islets, and has roles similar to those of the HNF family of transcription factors [44,45]. IPF1-MODY was first discovered in 1997 and is a very rare subtype of MODY [46]. Heterozygous mutation in the IPF1 gene causes ß-cell dysfunction and MODY, while homozygous mutation in the IPF1 gene results in neonatal diabetes [47].
Go to :
MODY6 (NEUROD1-MODY)
NEUROD1 encodes neurogenic differentiation 1, a basic helix-loop transcription factor involved in the development of endocrine cell lineage as well as neuronal development. Although heterozygous mutations in NEUROD1 result in MODY, homozygous mutations cause a novel syndrome of permanent neonatal diabetes and neurological abnormalities [48].
Go to :
MODY7 (KLF11-MODY)
The Krüppel-like factor (KLF) 11 gene is located on chromosome 2 and encodes a zinc-finger transcription factor. Mutations in KLF11 cause ß-cell dysfunction by modulating the expression of free radical scavengers. Two rare variants of KLF11 that impair its transcriptional activity (Ala347Ser and Thr220Met) were identified in families with early-onset type 2 diabetes [49].
Go to :
MODY8 (CEL-MODY)
The CEL gene encodes the bile salt-stimulated lipase, a major component of the pancreatic juice that is secreted by the pancreas into the digestive tract. The enzyme aids in the digestion of cholesterol and lipid-soluble vitamins, ester hydrolysis, and absorption of dietary fat from the intestine. In 2006, a heterozygous mutation in the CEL gene was identified in two families [50]. Exocrine pancreatic dysfunction (defined by fecal elastase deficiency) and ß-cell failure were found in a patient with single-base deletion in CEL. To date, heterozygous mutations in CEL have only been identified in three families [51].
Go to :
MODY9 (PAX4-MODY)
PAX4 is a homeodomain transcription factor that plays a central role in ß-cell development and function [52]. Two variants of PAX4 (R164W and IVS7-1G>A) were identified in two Thai probands [53].
Go to :
MODY10 (INS-MODY)
Dominant misfolding mutations in the INS gene are a common cause of isolated permanent neonatal diabetes; however, the age at which the disease develops can vary [54]. These mutations cause a severe folding defect, unfolded protein response, and ß-cell apoptosis.
Go to :
MODY11 (BLK-MODY)
B-lymphocyte kinase (BLK) is a nonreceptor tyrosine-kinase from the Src family of proto-oncogenes. It is expressed in ß-cells where it promotes insulin synthesis and secretion by up-regulating the transcription factors PDX1 and NKX6.1 [55]. These transcription factors enhance pancreatic ß-cell mass. Decreased BLK activity reduces the insulin content and renders the ß-cells less responsive to glucose, leading to decreased insulin secretion and, eventually, diabetes.
Go to :
MODY12 (ABCC8-MODY)
The ABCC8 gene encodes the sulfonylurea receptor 1 (SUR1) subunit of the pancreatic ß-cell ATP-sensitive potassium channel (KATP), which directly regulates insulin release. Recessive loss-of-function mutations in ABCC8 lead to the development of congenital hypoglycemic hyperinsulinism (CHI) [56], while dominantly inherited ABCC8 mutations may cause CHI with predisposition to insulin deficiency and diabetes later in life. Heterozygous activating mutations in ABCC8 cause MODY without a history of diabetes or hyperinsulinism in the neonatal period, and produce clinical manifestations similar to those of HNF1A/4A MODY [57]. Patients with mutations in ABCC8 respond to high-dose sulfonylurea therapy.
Go to :
MODY13 (KCNJ11-MODY)
KCNJ11 encodes the Kir6.2 subunit of the hetero-octameric KATP channel, which is highly expressed in pancreatic ß-cells. Homozygous or heterozygous mutations in this gene lead to the development of either transient or permanent neonatal diabetes within the first 6 months of life. Heterozygous KCNJ11 mutations were identified in 6 out of 96 families with early-onset type 2 diabetes [58]. Some of these carriers stopped the insulin therapy and switched to sulfonylurea.
Go to :
MODY14 (APPL1-MODY)
APPL1 (adaptor protein, phosphotyrosine interaction, PH domain, and leucine zipper containing 1) is the most recently identified MODY-related gene (first reported in 2015). APPL1 is an anchor protein with multiple functional domains that interact with other proteins, including the key components of the insulin-signaling pathway. Two loss-of-function mutations in the APPL1 gene have been identified in 60 families through whole-exome sequencing [9].
Go to :
Ways to avoid misdiagnosing patients with MODY
The diagnostic criteria for MODY are as follows: (1) presence of overt diabetes in at least three consecutive generations, with autosomal dominant mode of inheritance, (2) at least one family member diagnosed with diabetes before the age of 25, (3) absence of ß-cell autoantibodies, and (4) relatively preserved endogenous insulin secretion with a serum C-peptide level of >0.6 ng/mL. These diagnostic criteria can help discriminate MODY from type 1 and type 2 diabetes. Nevertheless, distinguishing MODY from type 1 or type 2 diabetes at presentation is often challenging [24,59,60].
Various algorithms have been developed to identify diabetic patients who should undergo genetic testing for MODY. Shields et al. [61] proposed a clinical prediction model to distinguish MODY from type 1 and type 2 diabetes. According to the model, patients with MODY have lower HbA1c levels than those with type 1 diabetes. Further, compared to type 1 diabetics, patients with MODY tend to have an older age at diagnosis and higher probability of being female and having a parent with diabetes. Compared to type 2 diabetics, patients with MODY tend to have a lower body mass index, lower HbA1c level, younger age at diagnosis, higher probability of being female and having a parent with diabetes, and lower probability of prior treatment with oral hypoglycemic agents or insulin. Although this model calculates a standardized probability of being diagnosed with MODY, it is important to bear in mind that the clinical manifestations of MODY may vary.
Sanger DNA sequencing, which is the gold standard for identifying mutations in MODY-related genes, might improve the chances of a correct diagnosis. However, genetic testing for MODY is expensive and may only be offered in specialist centers. Therefore, considerable efforts have been made to identify non-genetic biomarkers to facilitate the differential diagnosis of MODY. Persistent postprandial C-peptide level, which is measured in a spot urine sample, can discriminate HNF1/4A-MODY from type 1 diabetes [62]. Moreover, compared to type 2 diabetics, patients with HNF1A-MODY tend to have a lower level of high-sensitive C-reactive protein [63]. A proposed diagnostic algorithm for the identification of diabetic patients who might benefit from MODY genetic testing is presented in Fig. 1.
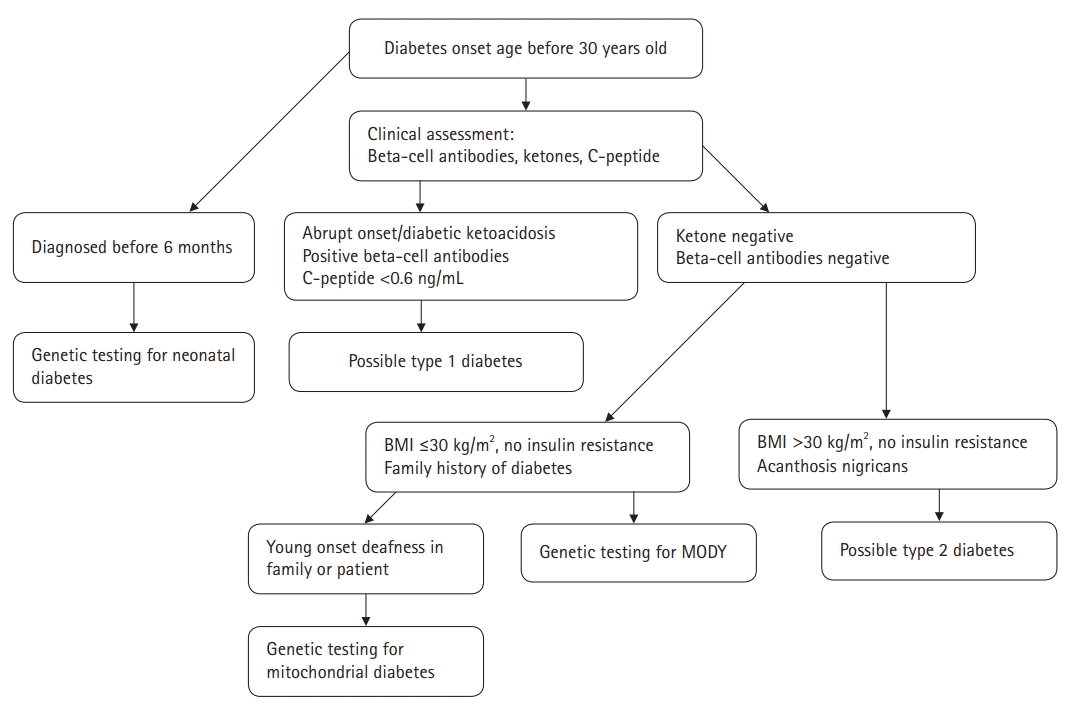
Go to :
Conclusion
The genetic etiology and pathophysiology of MODY have been widely researched. The biosynthesis and secretion of insulin from pancreatic beta cells are changed at various stages depending on the specific gene mutations (Fig. 2). In a recent study from Korea, 109 patients with clinically suspected MODY underwent targeted panel sequencing. The diagnosis was confirmed in 23 patients (21.1%) [64]. The diagnostic rate was similar to that in a large study on monogenic diabetes performed in the United Kingdom (27%) [14]. In the latter study, molecular genetic testing confirmed a diagnosis of GCK-MODY, i.e. the most common subtype of MODY, in 50% of the patients. This result was in agreement with that of studies in the Caucasian population [65,66]. In contrast, in a China-based study, HNF1A-MODY and GCK-MODY comprised only 9% and 1% of the total tested cases, respectively [67]. These results were similar to those obtained in Japanese studies [68-70]. A possible explanation for these discrepancies is that a large proportion of the MODY cases in China have defects in unknown MODY genes [67]. The studies from Korea, Japan, and China suggest that East Asia has a high prevalence of a not yet identified form of diabetes, i.e., ‘MODY X’ [64,67-72]. Next generation sequencing is one of the most powerful tools to discover unknown genetic defects [71,73], and attempts to identify new causative gene variants in MODY using whole-exome sequencing have been undertaken in Korea [72].
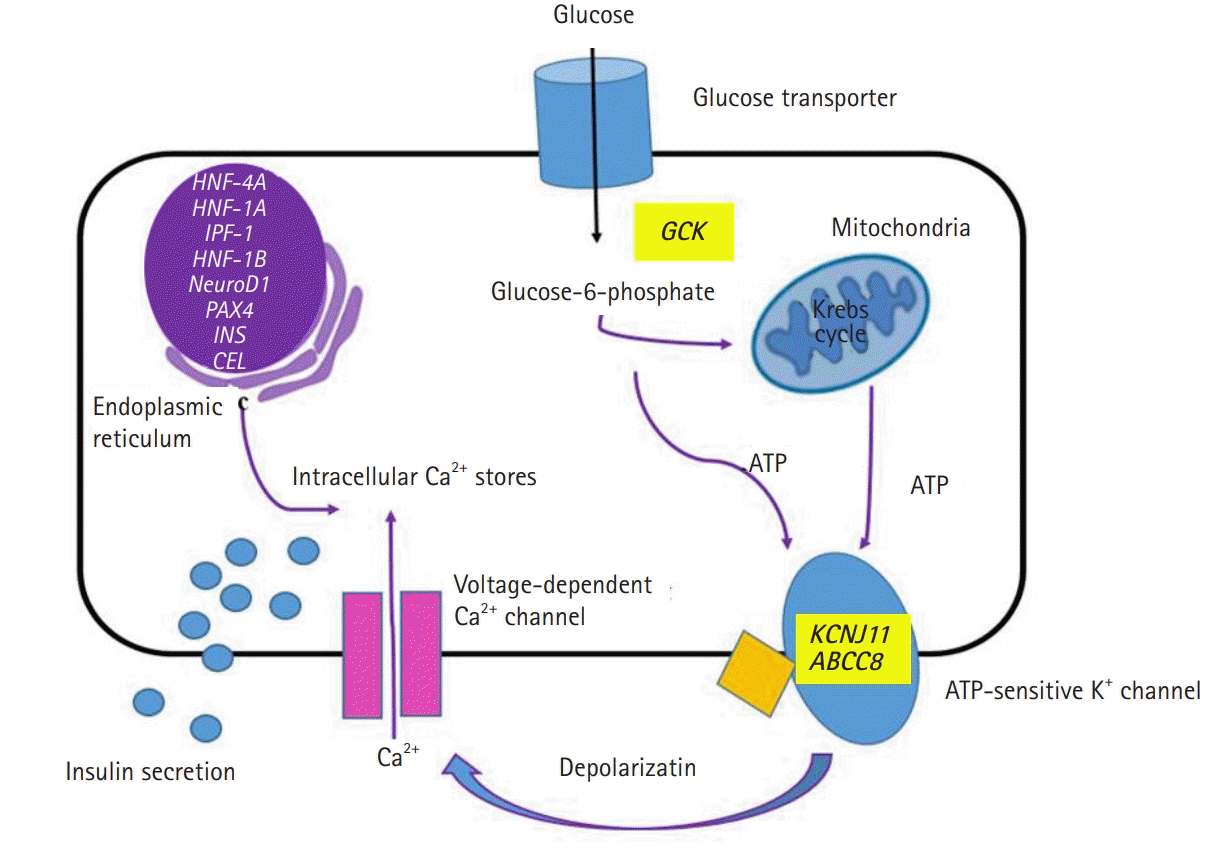
MODY is estimated to be the cause of 2%–5% of diabetes cases in Europe [11,12]. Owing to the increase in molecular genetic testing, the frequency of its detection has increased worldwide. Correct molecular diagnosis can help to ensure that patients with MODY receive optimal treatment. Metabolic profiling can also be an important diagnostic tool in patients with MODY.
Go to :
 XML Download
XML Download