

 PDF
PDF Citation
Citation Print
Print



Introduction
Diabetes mellitus (DM) is a metabolic disease that can cause complications in the peripheral nervous system and several organs in the body, including the kidney, eyes, and brain [1]. Among the complications of DM, cognitive dysfunctions are relatively less addressed. Some patients with type 1 and 2 DM present with cognitive dysfunctions. Both hypoglycemia and hyperglycemia are known to cause cognitive dysfunctions. However, the underlying pathophysiology is not well understood [2]. Research results about one hypothesis are conflicting, and studies with consistent results are not available. In addition, the most appropriate method for diagnosing, treating, and preventing cognitive dysfunctions in DM is not yet identified [2]. Although the mechanisms and results remain inconsistent, some patients with DM present with cognitive dysfunctions, and the decline in cognitive function has a significant impact on activities of daily living. Therefore, cognitive dysfunctions in patients with DM must be reviewed and summarized to obtain useful information. The present study aimed to review the characteristics of cognitive dysfunctions in patients with DM to summarize the factors affecting cognitive functions and the hypotheses about the mechanisms of cognitive dysfunctions.
Cognitive dysfunctions in type 1 diabetes mellitus
In a recent meta-analysis [3] that included 33 studies, the authors have analyzed the result of the cognitive function assessment performed during normal blood glucose state in adults with type 1 DM. Results showed that some cognitive domains, including speed of information processing, psychomotor efficiency, visual and sustained attention, mental flexibility, and visual perception, were significantly impaired in patients with type 1 DM compared with those of the controls. However, the cognitive domains of patients with type 1 DM were not significantly different from those of the controls, which include memory, motor speed, selective attention, and language. A recent systematic review [2] has shown that cognitive dysfunctions commonly observed in patients with type 1 DM are associated with the decreased speed of information processing, psychomotor efficiency, attention, memory, learning, problem solving skills, motor speed, vocabulary, visuoconstruction, visual perception, somatosensory examination, motor strength, mental flexibility, and executive function. Among these areas, decreased speed of information processing, psychomotor efficiency, attention, visuoconstruction, and mental flexibility have strong supporting data (Table 1).
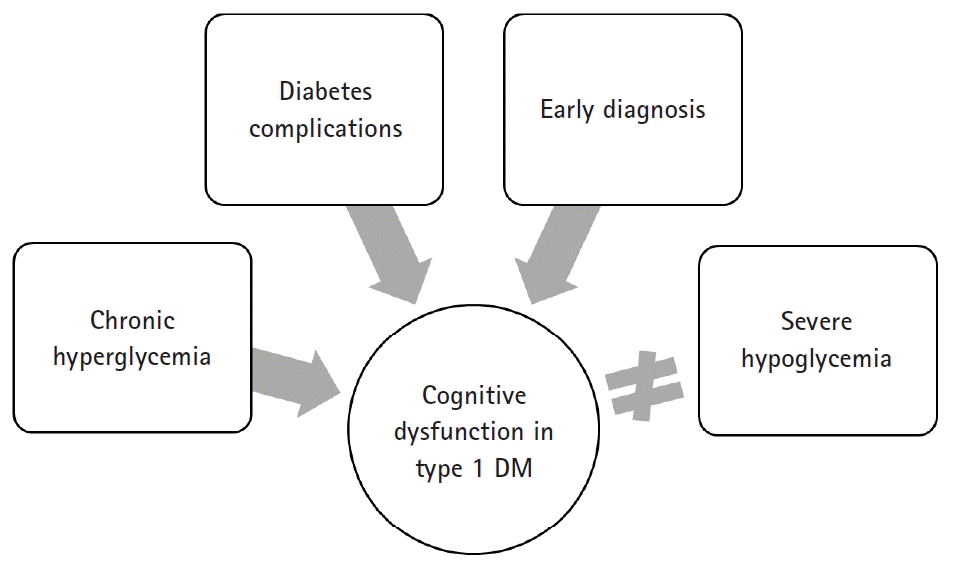
In the early 1990s, cognitive dysfunctions in type 1 DM were more prominent in individuals who repeatedly presented with severe hypoglycemia. This finding is consistent with those of anecdotal reports of severe hypoglycemia that leads to cortical changes in the frontal lobe and in other regions of the brain, such as the temporal lobe, basal ganglia, and hippocampus [4,5]. However, a more recent evidence indicated that hypoglycemic episodes may not cause cognitive dysfunctions. The results of longitudinal epidemiological studies have indicated that chronic hyperglycemia and microvascular complications cause DM-related cognitive dysfunctions. The Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications is a 18.5-year longitudinal epidemiological study with 1,144 participants. Of these participants, 40% had experienced one or more severe hypoglycemic episodes (blood glucose concentration <2.8 mmol/L accompanied by seizure or coma) during the study period. However, hypoglycemic episodes and cognitive dysfunctions were not significantly associated [6]. Rather, the 5 independent variables predicting the decrease in psychomotor speed over a follow-up period of 18.5 years were as follows: old age, low education, high lifetime hemoglobin A1c (HbA1c) concentrations, proliferative diabetic retinopathy, and renal complications [7]. In addition, an increase in carotid intima-media thickness was slightly associated with a decline in cognitive performance. In particular, retinopathy was most closely associated with cognitive dysfunctions in this study [6]. A limitation of this study is that only younger participants (aged <50 years) were included; thus, the study might have been conducted before the onset of cognitive dysfunctions due to hypoglycemia. However, this study showed that the risk of cognitive dysfunctions is lower, and the progression is also slower in young patients with DM who have good glycemic control. Systematic meta-analyses with similar results have reported that repeated hypoglycemia and cognitive decline were not correlated [3]. Several other studies have reported that microvascular complications are associated with an increased risk of cognitive decline [8].
However, hypoglycemia is associated with decreased cognitive function in the high-risk group, which was diagnosed early within the first few years of life [9,10]. Moreover, diagnosis at a younger age in patients with type 1 DM is associated with an increased risk of cognitive dysfunctions. Those who developed type 1 DM before the age of 4 years had impaired executive skills, attention, and processing speed compared to those diagnosed after the age of 4 years [11].
Appropriate glycemic control plays an important role in the cognitive functions of patients with type 1 DM. Better glycemic control improves functions, such as psychomotor efficiency, attention, motor speed, memory, and academic achievement [2]. In a DCCT study, patients with type 1 DM with a mean glycated hemoglobin (HbA1c) of 7.4% had significantly better motor speed and psychomotor efficiency than those with a mean HbA1c of 8.8% [6].
Cognitive function was worse in patients with type 1 DM who presented with DM complications. One study has reported that deficits in fluid intelligence, information processing, attention, and concentration are correlated to the presence of background retinopathy [12]. Periods of DM, proliferative retinopathy, macrovascular complications, and hypertension have been associated with decreased performance in psychomotor speed and visuoconstruction ability tests in patients with type 1 DM [13-15] (Fig. 1).
Several studies have reported that gender in patients with type 1 DM is associated with cognitive function. Skenazy and Bigler [16] have indicated that men with type 1 DM had decreased performance in measuring oscillation, strength grip, and somatosensory function than male controls, and the magnitude of this difference was greater than that between women with type 1 DM and gender-matched controls. A decline in verbal intelligence was observed in boys aged between 7 and 16 years with type 1 DM and was correlated with a deterioration in glycemic control. This was not observed in girls of the same age [17]. However, several studies have not observed gender differences. Future research with a better study design must be conducted.
Cognitive dysfunctions in type 2 diabetes mellitus
Numerous reports have shown that patients with type 2 DM are at increased risk of Alzheimer’s disease (AD) dementia and vascular dementia [18-22]. According to Bruce et al. [23], 17.5% of elderly patients with type 2 DM present with moderate to severe deficits in activities of daily living; 11.3% with cognitive impairment; and 14.2% with depression. A recent systematic review [2] has shown that cognitive dysfunctions commonly reported in patients with type 2 DM are associated with memory, psychomotor speed, executive function, processing speed, complex motor function, verbal fluency, and attention. Among these areas, memory, psychomotor speed, and executive function have strong supporting data (Table 1). By contrast, in some studies, the effects of subtle neurocognitive impairment on the daily activities of patients with type 2 DM are not fully elucidated, and there is a debate on whether such condition is attributed to changes in diabetic brain function [24]. In particular, the problem in daily function observed in patients with type 2 DM may be due to depression [23].
Proper glycemic control plays an important role in the cognitive function of patients with type 2 DM. In nearly 2,000 postmenopausal women, those with an HbA1c ≥7.0% had a four-fold increased risk of developing mild cognitive impairment [25]. Grodstein et al. [26] have found that elderly participants who were receiving oral diabetic medications, other than insulin, had similar cognitive scores with participants without DM. Other studies have shown an inverse relationship between HbAlc and working memory [27,28], executive function [27], learning [29], and complex psychomotor performance [29,30] in patients with type 2 DM, thereby supporting the hypothesis that worsening glycemic control leads to cognitive dysfunctions similar to those observed in type 1 DM.
Nevertheless, the benefits of aggressive glucose management in type 2 DM are not fully elucidated [31-33]. The target value of HbA1c in intensive therapy in the study was approximately <6.0% or >1.5% lower than that in the standard therapy (targeting level of 7.0–7.9%). However, several studies have shown that intensive glycemic control did not have a positive effect on cognitive function [31-34]. The Memory in Diabetes substudy of the Action to Control Cardiovascular Risk in Diabetes trial has reported that aggressive glucose-lowering therapy did not have positive effects on not only cognitive function but also total brain volume during the 40-month follow-up in patients with type 2 DM [34].
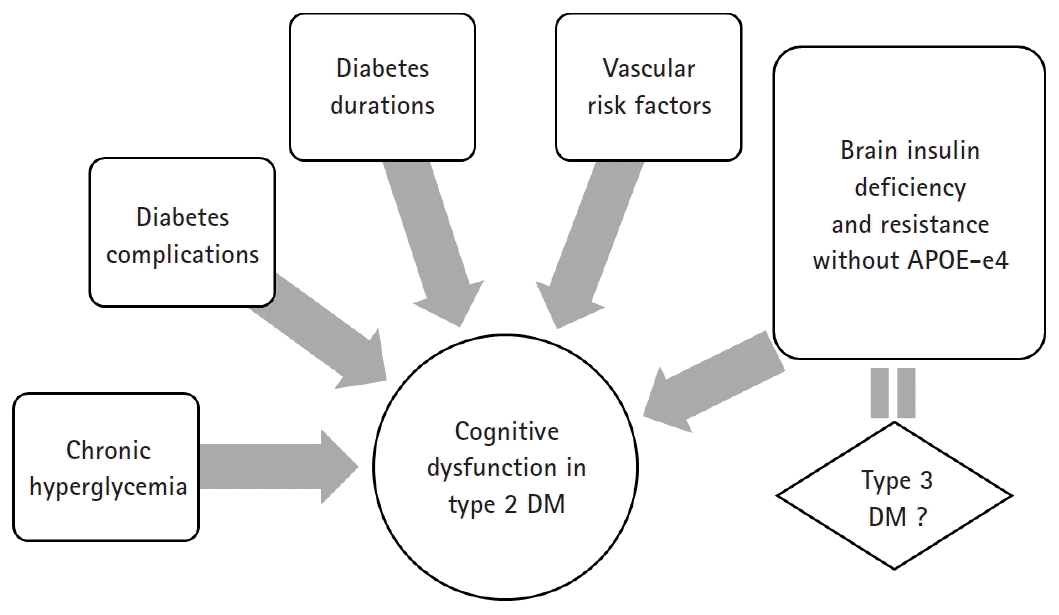
Similar to type 1 DM, type 2 DM is associated with deficits in cognitive function when accompanied by diabetic complications, such as peripheral neuropathy [28,35]. One study has reported that diabetic retinopathy in men, but not in women, was associated with a decrease in cognitive performance [28]. The duration of type 2 DM and chronic hyperglycemia were associated with changes in cognitive function [8,36] (Fig. 2).
Some reports have shown that impaired glucose tolerance before DM was a risk factor of cognitive decline [37]. Numerous studies have shown that patients with impaired glucose tolerance had lower Mini-Mental State Examination and long-term memory scores [36], decreased verbal fluency [38], and worse Alzheimer’s dementia [20] and vascular dementia [19] than control subjects. However, not all studies obtained similar results.
Pathophysiology of cognitive dysfunction in diabetes mellitus
Thus far, the underlying pathophysiology of cognitive dysfunctions in DM is not well understood. However, several hypotheses have been proposed, and research results that validate such hypotheses have been published.
First, there is a hypothesis that cognitive dysfunction is correlated to hyperglycemia. Although hyperglycemia may be associated with decreased cognitive function in type 1 and 2 DM, the mechanism by which hyperglycemia mediates is still unclear. In other organs, hyperglycemia affects function by the following mechanism: polyol pathway activation, increased formation of advanced glycation end products, diacylglycerol activation of protein kinase C, and increased glucose shunting in the hexosamine pathway [39-42]. Several studies have indicated that the same mechanisms may work in the brain.
Second, there is a hypothesis that vascular disease plays an important role. Patients with DM are 2–6 times more likely at risk of thrombosis, which contributes to cognitive dysfunction [43-45]. Thickening of capillary basement membranes (the hallmark of diabetic microangiopathy) is also found in the brain of patients with DM [46]. The duration of illness was associated with the decreased global rates of cerebral blood flow in patients with DM. Interestingly, the cerebral blood flow rate of patients with DM is similar to that observed in patients with AD [47]. The coexistence of ischemia and hyperglycemia was considered detrimental to the brain. One potential mechanism by which hyperglycemia may enhance ischemic damage is lactate accumulation [48]. Hyperglycemia produces more substrate for the formation of lactic acid, causing cellular acidosis and exacerbated damage. Another mechanism is glutamate accumulation in the context of hyperglycemia and ischemia [49,50].
Third, whether hypoglycemic episodes contribute to cognitive impairment is controversial, and it may depend on the patient’s age. However, extremely long-term severe hypoglycemia causes brain damage and even death [45]. In animal models, blood glucose levels of 0.12–1.36 mmol/L for 30–60 min lead to increased extracellular aspartate levels, alkalemia, and nerve necrosis with nerve energy damage, thereby resulting in a flat electroencephalograph result [51]. The cortex, basal ganglia, and hippocampus may be most vulnerable to hypoglycemia, and autopsies performed in human patients who died from hypoglycemia showed lamellar necrosis and gliosis in these areas [52]. Other human autopsy studies conducted after death secondary to hypoglycemia have shown multifocal or diffuse necrosis of the cerebral cortex and chromatolysis of ganglion cells [53]. Some researchers have hypothesized that hypoglycemia-induced neuronal damage occurs due to the overactivation of the excitatory neurotransmitter subtypes of the N-methyl-D-aspartate receptor (NMDA) receptor [54]. Interestingly, NMDA receptor antagonists prevent neuronal necrosis, indicating a potential treatment for hypoglycemia-induced brain injury [55].
Fourth, in recent studies, insulin resistance has been considered a mechanism of cognitive decline in patients with DM. In fact, historically, the brain was believed to be an insulin-independent organ. However, recent studies have questioned such concept. There is a growing body of evidence showing that insulin resistance can play an important role in the pathogenesis of AD. Since glucose is the main fuel of the brain, the brain is starved due to the lack of glucose uptake and utilization. Insulin stimulates brain glucose uptake and utilization, metabolism, memory, and cognition. Insulin resistance/deficiency induces impairments in glucose metabolism and disrupts brain energy balance, thereby increasing oxidative stress, production of reactive oxygen species, deoxyribonucleic acid damage, and mitochondrial dysfunction, all of which drive pro-apoptosis, pro-inflammatory, and pro-amyloid-beta (Aβ) cascades [56]. Therefore, insulin and insulin-like growth factor (IGF) signaling pathways play critical roles in brain functions, such as cognitive functions in the central nervous system (CNS). Insulin, IGF-1 and IGF-2 polypeptides, and receptor genes are expressed in neuronal and glial cells throughout the brain. Their highest levels are in structures that are strongly targeted by neurodegeneration, particularly in AD [56-58]. Correspondingly, experimental depletion or suppression of brain insulin receptor expression and function causes cognitive dysfunction and the molecular and biochemical abnormalities observed in AD [59]. In this context, it is argued that AD is considered a brain disease with complex features of type 1 (insulin deficiency) and type 2 (insulin resistance) DM. To consolidate this concept, numerous investigators have proposed that AD should be referred to as type 3 DM [60,61].
Evidence of type 3 diabetes mellitus in Alzheimer’s disease
Sporadic AD as the cause of Aβ accumulation, which accounts for more than 90% of AD cases, is not yet well understood. Recent evidence has indicated that insulin/IGF resistance in the brain as both a cause and outcome. Insulin stimulation promotes the transport of Aβ from the trans-Golgi network from which it is derived to the plasma membrane for extracellular secretion [62]. Furthermore, insulin inhibits intracellular accumulation of Aβ and degradation by insulin-degrading enzymes [63,64]. Impaired insulin signaling disrupts amyloid-beta precursor protein peptides (AβPP) processing and AβPP-Aβ clearance in the brain [65].
The tau pathology is a representative neuropathological finding of AD along with Aβ. Moreover, studies have shown that insulin resistance affects tau expression and phosphorylation. Tau expression and phosphorylation are regulated by insulin and IGF [66,67]. In AD, brain insulin and IGF resistance impair signaling through phosphoinositol-3-kinase (PI3K), Akt, and Wnt/β-catenin and promote glycogen synthase kinase-3 beta (GSK-3β) activation. Overactivation of GSK-3β is responsible for tau hyperphosphorylation, which promotes tau misfolding and fibril aggregation.
The strongest evidence supporting the notion of type 3 DM in AD was from an experimental study of rats that received streptozotocin, a pro-diabetic drug, via intracerebroventricular injection. Streptozotocin-treated rats develop cognitive dysfunctions as characterized by lack of spatial learning and memory, brain insulin resistance and deficiency, and AD-like neurodegeneration [68,69]. Targeted exposure to pro-diabetic agents can cause neurodegeneration with structural, molecular, biochemical, and functional abnormalities that closely mimic the pathology of AD in humans. In particular, streptozotocin is a nitrosamine-related compound that can be found in processed and preserved foods, and it causes cognitive impairment, AD-type neurodegeneration, and brain insulin resistance with experimental exposures to low, sub-mutagenic doses. Some authors have shown that over the last few decades, western populations have been constantly presenting with increasing levels of exposure to nitrosamines due to constant and gradual contact with nitrates and nitrites in the environment, agriculture, and food resources, and this has recently been linked to the increased incidence of dementia [68,69].
Some studies have indicated that insulin resistance affects neurotransmission and memory formation. Streptozotocin-induced diabetic rats have reduced acetylcholine production and release as compared to control rats [70]. A mouse model in which cholinergic activity is blocked by scopolamine experienced memory loss and hyperactivity, a deficit that may be reversed by glucose administration [71]. Moreover, glucose administration with elevated endogenous insulin levels or insulin administration to patients with AD has also been shown to cause changes in behavior, possibly by enhancing cholinergic activity [72]. DM and insulin may affect long-term potentiation in the opposite way. Long-term potentiation is important for memory formation and is induced by NMDA receptor activation, a process that is upregulated in the presence of insulin. However, rats presumed to have DM and relative insulin deficiency has reduced long-term potentiation in the hippocampus as measured via electrophysiology [73,74]. Thus, brain insulin resistance in AD may require a higher level of insulin to stimulate memory [75]. However, it is unclear whether the direct effect of insulin is directly or indirectly influenced by other mediators [76].
Recently, a noteworthy report has indicated that the relationship between insulin resistance and cognitive impairment in AD is dependent on the presence or absence of the apolipoprotein E (APOE)-e4 allele. The presence of the APOE-e4 allele itself increases the risk of AD by a factor of 2–8, which plays a rather opposite role in the development of AD associated with insulin resistance. Insulin resistance is only an important risk factor of AD in patients without the APOE-e4 allele [20]. Participants with AD without the APOE-e4 allele also had improved memory scores in the hyperinsulinemic setting, which was not the case for APOE-e4 allele-positive participants [77] (Fig. 2).
One of the important reasons for the active research in these fields may be the anticipation that antidiabetic drugs may play a preventive or therapeutic role in AD, which is known to have no cure. It has been hypothesized that neurodegeneration and cognitive dysfunctions in AD may be reduced or prevented with the early treatment of insulin-sensitizer antidiabetic agents, such as peroxisome proliferator-activated receptor (PPAR) agonists. Studies have reported that the PPAR agonist treatments prevent cerebral atrophy, preserve insulin and IGF-2 receptor-bearing CNS neurons, and specifically prevent it from ic-streptozotocin-induced deficits in learning and memory [78]. Some trials have shown that rosiglitazone, a PPAR-gamma, have a beneficial effect on memory in patients with AD. In a small randomized study published by Watson et al. [79] in 2005, patients with mild AD who were treated with rosiglitazone for 6 months had better memory and selective attention than the controls. A larger study published in 2006 has shown improvement in cognitive function after the administration of rosiglitazone for 6 months in patients with AD without the APOE-e4 allele. However, no improvements were observed in patients with AD with the APOE-e4 allele [80]. Similar studies have been conducted. However, the results remained inconsistent reports. The results of the systematic review and network meta-analysis [81] in 2018 have shown the significant effects of antidiabetic agents on AD and mild cognitive impairment, and pioglitazone 15–30 mg was more effective than placebo in a network meta‐analysis.
Conclusion
Several studies about cognitive dysfunctions in patients with type 1 and type 2 DM have been conducted, and the results were inconsistent. Although some studies obtained conclusions, limitations were still observed. Type 1 DM has been associated with a decrease in the speed of information processing, psychomotor efficiency, attention, mental flexibility, and visual perception, and type 2 DM has been associated with memory deficits, a decrease in psychomotor speed, and reduced frontal lobe/executive function. Improved glycemic control and decreased diabetic complications may be associated with the prevention of cognitive dysfunction. However, the benefits of intensive glucose control in type 2 DM remain unclear. Severe hypoglycemic episodes may not have a long-term cognitive impact. However, it may cause cognitive impairment in high-risk patients diagnosed with DM (<4 years). The underlying pathophysiology is not well understood. Some authors have claimed that AD is type 3 DM with a combination of insulin deficiency and resistance particularly in patients with sporadic AD without the APOEe4 allele. However, results of previous studies remained inconsistent, and more consistent results must be obtained. If a patient reports that their performance is worsening or if they ask about the effects of DM on functioning, screening and assessment are recommended.
 XML Download
XML Download