

 PDF
PDF Citation
Citation Print
Print



Introduction
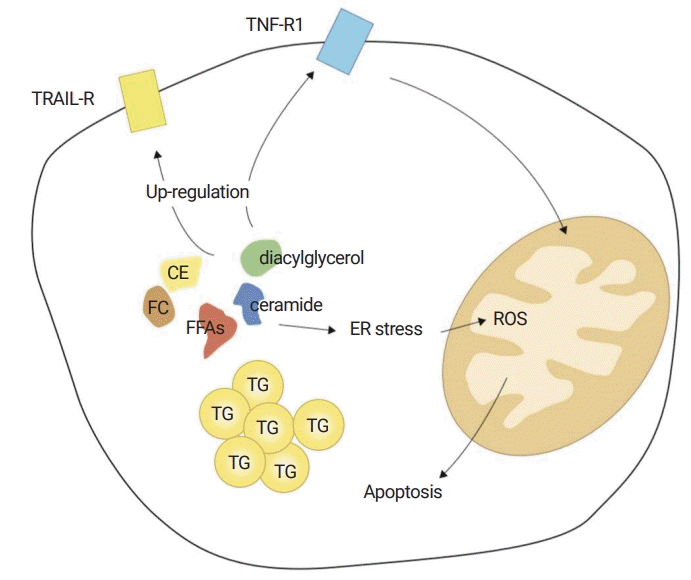
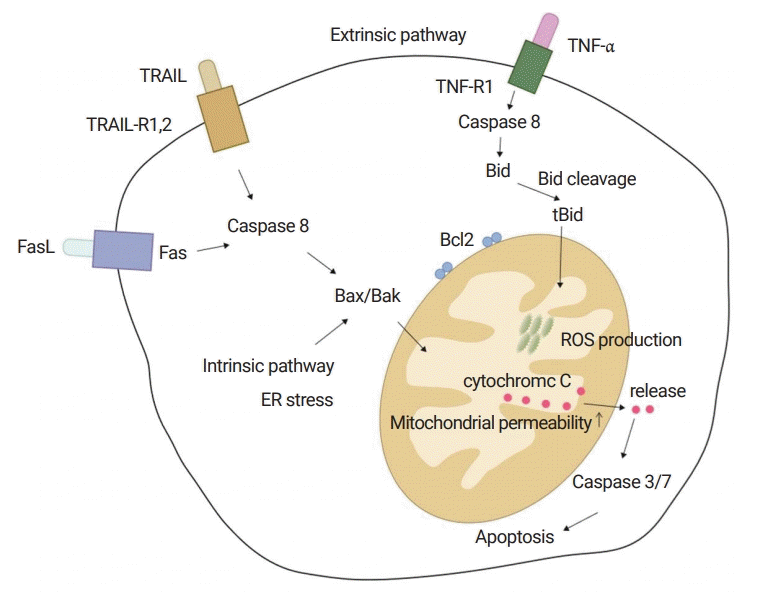
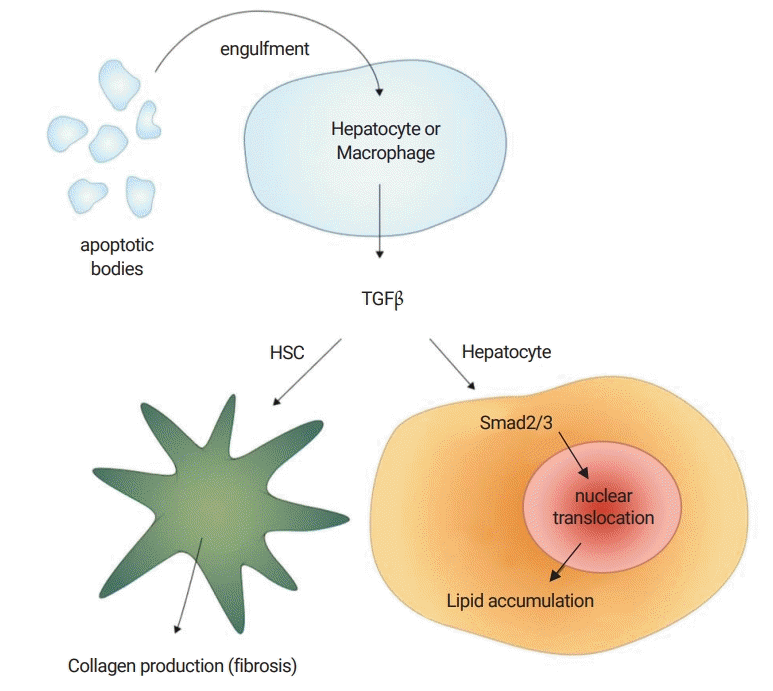
Nonalcoholic fatty liver disease (NAFLD) is a chronic disease in which lipid accumulates in hepatocytes, and hepatocyte ballooning is seen pathologically. The spectrum is wide, ranging from simple steatosis to steatohepatitis (NASH), fibrosis, and even cirrhosis [1]. Lipotoxicity is the main pathophysiology associated with NASH initiation and progression [2]. Even though the main lipid is a form of triglyceride (TG), other lipid metabolites also accumulate. Lipid metabolites, such as free cholesterol (FC) and free fatty acids (FFAs), cause apoptosis via up-regulation of the death receptors, sensitizing them to inflammatory cytokines [2] (Fig. 1). Caspases and the Bcl2 families are involved in the apoptotic pathway [3] (Fig. 2). Even hepatocytes, including Kupffer cells, phagocytize apoptotic cell debris. They release transforming growth factor-β (TGF-β), the main cytokine of fibrosis, which causes the activation of hepatic stellate cells (HSCs) and more accumulation of lipids in the hepatocytes via Smad2/3 [4] (Fig. 3). Therefore, lipotoxicity and apoptosis may be suitable therapeutic targets for the treatment of NASH.
Endotoxin (lipopolysaccharide) activates intrahepatic macrophages (Kupffer cells) via toll like receptor 4 (TLR4), which then release inflammatory cytokines including C-C motif chemokine ligand 2 (CCL2). C-C motif chemokine receptor 2 (CCR2)-positive bone marrow (BM)-derived monocytes and HSCs respond to CCL2 and are recruited to the liver [6]. CCR5 is expressed on HSCs and lymphocytes. CCR5 cells infiltration via chemokine-chemokine interaction cause inflammation and fibrosis in the liver [7]. The phase 2b CENTAUR trial was performed to evaluate the efficacy of cenicriviroc (CVC), a dual inhibitor of CCR2/CCR5 [8].
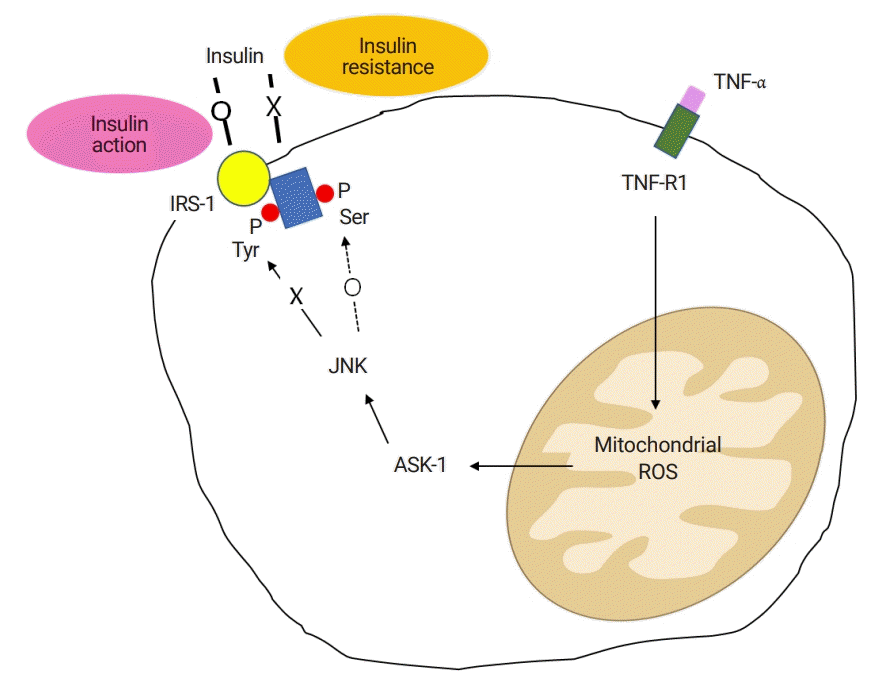
Obesity and insulin resistance are the main risk factor for NASH progression, as well as for cardiovascular disease [9,10]. Hyperglycemia and hyperinsulinemia increase the expression of connective tissue growth factor and activate the type 1 collagen gene in HSCs [9]. Hyperglycemia and hyperinsulinemia have been associated with hepatocellular carcinoma (HCC) in NASH [10]. Insulin sensitizers, such as thiazolidinediones and metformin, are prescribed for the treatment of type 2 diabetes worldwide [11]. Accordingly, they have been investigated as treatments for NASH [12]. Apoptosis signal-regulating kinase 1 (ASK1) is a key enzyme in insulin resistance and inflammation in NASH. The mitochondrial oxidative stress caused by inflammatory cytokines activates c-Jun-N-terminal kinase (JNK) via ASK1 [13]. The activated JNK interrupts insulin action by serine phosphorylation of the insulin receptor substrate 1, not tyrosine phosphorylation [14] (Fig. 4). Selonsertib, an ASK1 inhibitor, improved metabolic parameters in phase 2 clinical trials [14]. Two phase 3 trials are ongoing [13,14].
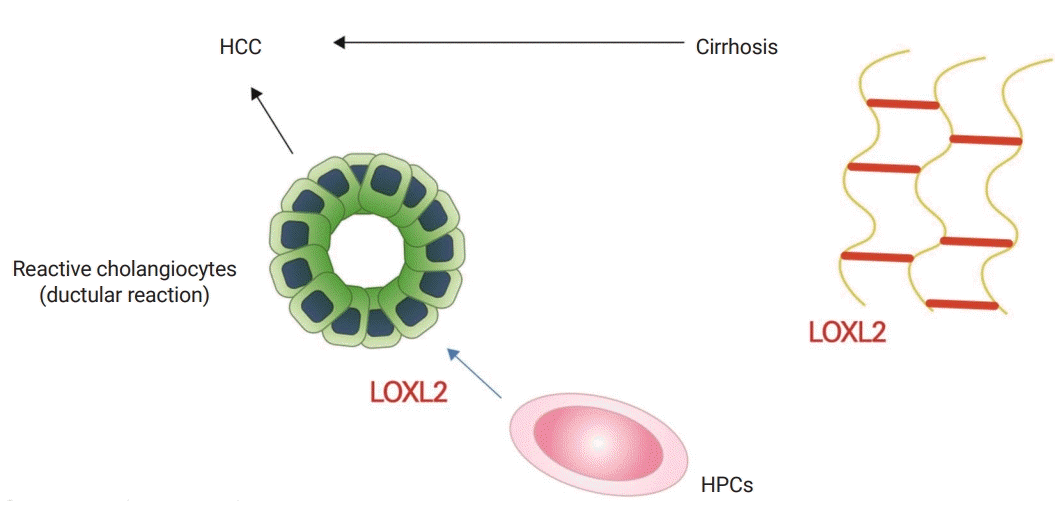
Cirrhosis is the final event in the fibrotic process. Lysyl oxidase-like 2 (LOXL2) is an enzyme which stabilizes collagen crosslinking. LOXL2 is involved in fibrosis progression, cirrhosis, and even HCC development by directing hepatic progenitor cells toward cholangiocyte differentiation (ductular reaction) [15] (Fig. 5). Simtuzumab, an anti-LOXL2 monoclonal antibody, was tried in clinical trials anticipating an anti-fibrotic effect [16].
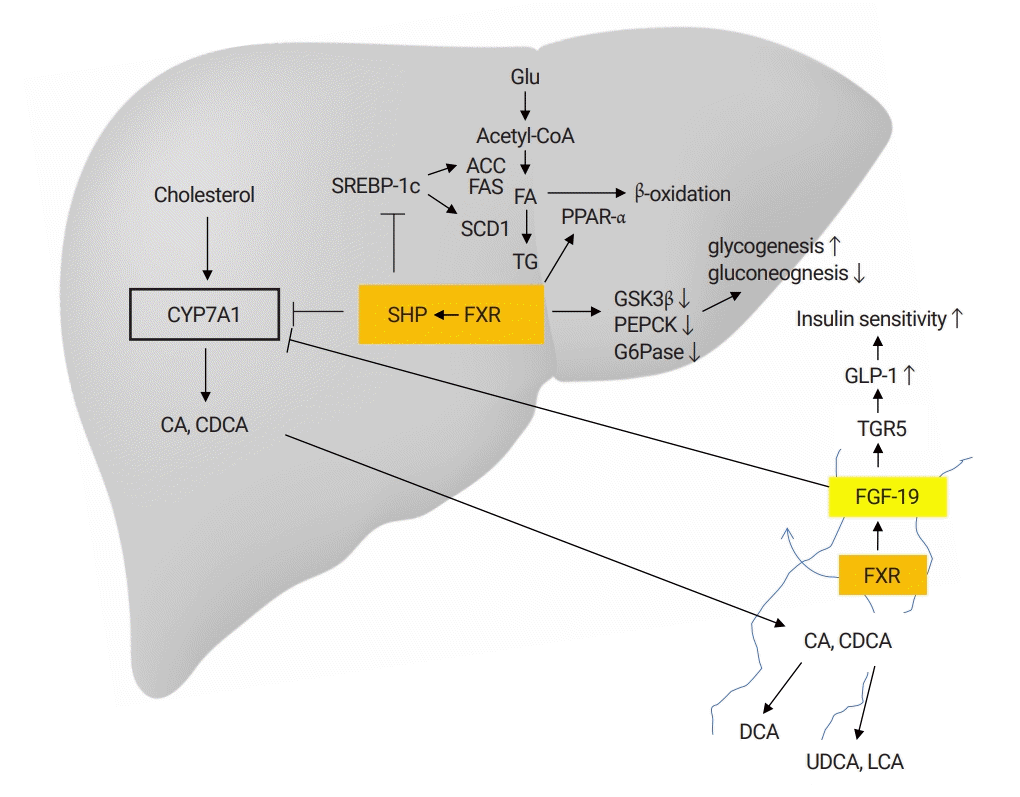
Recently, the farnesoid X receptor (FXR), a bile acid synthesis regulator, has gained attention. In the ileum, FXR activates fibroblast growth factor 19 (FGF-19), which increases insulin sensitivity by glucagon-like peptide-1 (GLP-1) activation. Moreover, in the liver, FXR inhibits TG synthesis by inhibiting the sterol regulatory element-binding protein 1c (SREBP-1c) via the short heterodimer partner (SHP), stimulates the β-oxidation of FFAs by peroxisome proliferator-activated receptors (PPAR)-α activation, and improves glucose homeostasis [17] (Fig. 6). Obeticholic acid (OCA), an FXR agonist, has been tested for the treatment of NASH [18].
Although many drugs are under phase 2 or 3 clinical trials, none have been approved by the Food and Drug Administration (FDA) to date. This review will cover the basic principles of pharmacologic therapy in practice, and the important results of phase 2 or 3 clinical drug trials, focusing on pathophysiology in detail.
Basic principles of pharmacologic therapy
To treat underlying associated diseases, such as type 2 diabetes, dyslipidemia, and hypertension.
1. The use of anti-diabetic drugs in cases of co-morbid type 2 diabetes
1) Pioglitazone and rosiglitazone
Pioglitazone and rosiglitazone are anti-diabetic drugs of the thiazolidinedione family which stimulate PPAR-γ, decreasing fatty acid migration into the liver and increasing β-oxidation by AMP-activated protein kinase (AMPK) activation [19]. Moreover, PPAR-γ inhibits HSC activation and increases adiponectin levels [19,20]. Randomized controlled trials (RCTs) were conducted based on this background [21]. Insulin resistance and liver enzymes improved during the treatment, but the effect was not sustained after the treatment was stopped. Histologic improvement in steatosis, inflammation, and hepatocellular ballooning was observed. However, fibrosis improvement was not confirmed. Common adverse effects were weight gain and edema. Safety concerns regarding myocardial infarction and bladder cancer remain [21].
2) Metformin
Metformin is a first-line anti-diabetic drug of the biguanide family. Metformin attenuates insulin resistance via activation of the AMPK pathway, hence it increases the uptake of peripheral glucose into the liver, muscle, and adipose tissue. It also suppresses gluconeogenesis in the liver and lipolysis in adipose tissue [21]. A study on metformin showed that the liver enzyme normalization and histologic improvement rates were higher compared to the vitamin E or diet-only groups [22]. Metformin showed a weight-reducing effect and a satisfactory safety profile. Some studies showed the improvement of insulin resistance and aminotransferase levels. However, a recent meta-analysis concluded that metformin did not improve liver histology. Therefore, it is not recommended as first-line therapy for the treatment of NASH patients without diabetes [23].
3) Glucagon-like peptide 1 agonist
The native GLP-1 agonist increases insulin secretion and suppresses glucagon secretion. It also delays gastric emptying and decreases appetite. However, endogenous GLP-1 is degraded in minutes by dipeptidyl peptidase 4 [24]. Liraglutide, a long-acting GLP-1 agonist, was approved in 2009 as an anti-diabetic drug in obese patients by the effects of weight reduction, pancreatic beta-cell function improvement, HbA1c reduction, and beneficial effects on blood pressure. In the LEAN study, liraglutide showed NASH resolution in 39% (9/23) of the patients, weight reduction and glucose control [24]. In December 2014, liraglutide (Saxenda®) was approved by the FDA for the treatment of obesity (body mass index [BMI] ≥30, or BMI ≥27 plus dyslipidemia or type 2 diabetes or hypertension) based on the SCALE study [25]. All GLP-1 agonists are given as injections.
4) Sodium/glucose cotransporter 2 inhibitor
SGLT2 inhibitors are anti-diabetic drugs which act by increasing the urinary excretion of glucose. They also reduce body weight and blood pressure [26]. Several pilot studies have shown significant reductions in alanine transaminase (ALT), body weight, and the fatty liver index in NAFLD patients. The impact on liver histology was not confirmed [27]. A recent RCT showed that luseogliflozin significantly reduced liver fat deposition, as well as visceral fat, HbA1c, and BMI compared to metformin [28]. In another RCT, ipragliflozin showed body weight and visceral fat reduction compared to pioglitazone [29]. A few pilot studies of SGLT2 inhibitors are ongoing [26].
2. The use of anti-hyperlipidemic agents in cases of dyslipidemia
1) Statins
As mentioned in the introduction section, lipotoxicity is the main pathophysiology of NASH initiation and progression. The altered quality of lipids, rather than their quantity, causes lipotoxicity [2]. Altered lipid metabolites up-regulate death receptors, sensitizing them to inflammatory cytokines. They also cause endoplasmic reticulum (ER) stress. Extrinsic and intrinsic stimuli cause mitochondrial oxidative stress, then finally, apoptosis. Moreover, FC accumulation in the HSCs increases TLR4, then activates TGF-β via bone morphogenic protein and activin membrane-bound inhibitor inhibition, which causes a vicious cycle of FC accumulation [30].
Statins have lipid-lowering effects as HMG-CoA reductase inhibitors. They also have anti-oxidant and anti-inflammatory effects [2]. Atorvastatin has been reported to decrease mitochondrial FC and increase glutathione levels [2]. These agents should be considered for the prevention of cardiovascular diseases in NAFLD patients with hypercholesterolemia. Several studies have suggested that statins may improve liver enzymes and histology in patients with NASH. However, no RCTs with histological endpoints have been conducted. Until RCTs with histological endpoints are undertaken, statins are not recommended for the treatment of NASH without dyslipidemia [23].
2) Omega-3 fatty acids
N-6 fatty acids have inflammatory, and n-3 fatty acids have anti-inflammatory action. Moreover, an increased n-6/n-3 fatty acids ratio increased the HCC risk in a NASH mouse model [31]. Omega-3 fatty acids showed improvement in hepatic steatosis, insulin sensitivity, oxidative stress, and anti-inflammatory action in an animal model [32]. Omega-3 fatty acids are currently approved in the United States to treat hypertriglyceridemia [23]. Two large studies failed to show therapeutic benefit in patients with NAFLD/NASH [33,34]. Therefore, they are not recommended for the treatment of NAFLD without hypertriglyceridemia [23].
3) Fibrates
Fibrates, such as bezafibrate or fenofibrate, are extensively used for the treatment of hypertriglyceridemia. But there is little data on NAFLD/NASH.
4) Ezetimibe
Ezetimibe inhibits the intestinal absorption of luminal cholesterol by binding to the Niemann-Pick C1-like 1 transporter in the membrane of the enterocyte brush border. In an animal model, it reduced cholesterol absorption up to 15 to 20% and hepatic fat content, and improved insulin resistance [35]. But the MOZART RCT trial failed to demonstrate reductions in liver fat contents or an improvement in the liver histology of NASH patients [36].
3. Angiotensin receptor blockers in cases of co-morbid hypertension
Angiotensin II type I receptors are expressed on activated HSCs. Therefore, angiotensin receptor blocker (ARBs) may have anti-fibrotic effects in NASH. In an animal study, olmesartan improved fibrosis in NASH [37]. In a study of 54 NASH patients, telmisartan improved insulin resistance and NASH scores compared to valsartan [38]. A losartan study in NASH patients failed because of slow recruitment of patients due to the already-widespread use of ARB in NASH patients [39].
New drugs under clinical trial
OCA, selonsertib, CVC, and elafibranor are in phase 3 trials. Emricasan, aramchol, simtuzumab, NGM282, and BMS-986036 trials have been completed or are in phase 2 trials [40].
1. Farnesoid X receptor agonist: obeticholic acid
OCA is a first-in-class selective FXR agonist. Its mechanism of action scheme is shown in Fig. 6. In an animal model, OCA improved insulin sensitivity, glucose and lipid metabolism, and showed anti-inflammatory and anti-fibrotic effects in hepatic, renal, and intestinal tissues [41]. Two major phase 2 clinical trials were conducted to evaluate the safety and efficacy of OCA in biopsy-proven NASH patients [41,42]. In the FLINT phase 2b trial (n=283), 45% of the patients in the 72-week OCA group achieved the primary outcome (a decrease in NAFLD activity score by ≥2 points without worsening of fibrosis) compared to 21% in the placebo group (p=0.0002). Fibrosis improvement was observed in 22% of the patients in the OCA group compared to 13% in the placebo group (p=0.08). The main adverse events of OCA were increased low-density lipoprotein (LDL) cholesterol, decreased high-density lipoprotein cholesterol and pruritus (23%) [42]. The CONTROL trial (NCT02633956) combining statins is ongoing. Two international phase 3 trials, RENERATE (NCT02548351) and REVERSE (NCT03439254) are now ongoing [41].
2. ASK1 inhibitor: selonsertib
ASK1 activation by mitochondrial oxidative stress, then JNK activation, is an important process in insulin resistance and inflammation [43]. The schematic mechanism of action is shown in Fig. 4. Selonsertib, an ASK1 inhibitor, significantly improved, not only metabolic parameters but also histologic parameters, such as hepatic steatosis, inflammation, and fibrosis [14]. In a 24-week clinical trial with or without simtuzumab, 43% (13/30) of the patients in the 18 mg selonsertib group showed reduced fibrosis (≥one stage) compared with 30% (8/27) in the 6 mg selonsertib group and 20% (2/10) in simtuzumab-alone group [14]. Two phase 3 clinical trials, STELLAR-3 (NCT03053050) and STELLAR-4 (NCT03053063), are now ongoing [40]. Common adverse effects are headache, nausea, fatigue, and upper abdominal pain [14].
3. CCR2/CCR5 dual inhibitor: cenicriviroc
The role of CCR2 and CCR5 for NASH progression are described in the introduction section. CVC is a potent inhibitor of CCR2/CCR5. In the phase 2b CENTAUR trial (n=289), CVC did not meet the primary endpoint (≥2-point NAS improvement or NASH resolution) but showed ≥ one stage fibrosis improvement after 1 year of treatment (20% vs. 10% in the placebo group, p=0.023). In contrast, CVC did not change body weight, aminotransferase levels, or insulin resistance. It was safe and tolerable, especially in terms of infection concerns [8]. The large phase 3 AURORA trial (NCT03028740) was initiated based on the efficacy and safety data of the CENTAUR trial [7,40]. About 2,000 patients were randomized 2:1 (CVC 150 mg or placebo) to evaluate liver fibrosis improvement. They will undergo three consecutive liver biopsies (baseline, after 1 and 5 years). The trial will end in 2019. FDA approval might be determined by the outcome of the trial [40].
4. PPAR α/δ agonist: elafibranor (GFT505)
PPARs are composed of three isoforms: α, β/δ, and γ. PPARs are expressed in many tissues but differently distributed between the isoforms. For example, the α isoform is mainly in the liver and skeletal muscles, while the δ isoform is found in all tissues. PPARs participate in fatty acid oxidation and energy balance. Elafibranor (GFT505), a dual PPAR α/δ agonist, improved plasma lipids and glucose homeostasis, insulin resistance, and reduced liver inflammatory markers [44]. The phase 2b GOLDEN-505 trial (NCT0164849) was conducted to evaluate the safety and efficacy of elafibranor. A total of 276 NASH patients without cirrhosis were randomized to three groups (120 mg, 80 mg, and placebo). The primary endpoint (NASH reversal without progression of fibrosis at 52 weeks) was not met but post-hoc analysis based on a modified definition of response (disappearance of ballooning with the disappearance or mild persistence of lobular inflammation and no worsening of fibrosis), showed significant superiority in the 120 mg group. Liver enzymes, LDL-cholesterol, HbA1c, and inflammatory markers were significantly reduced in the 120 mg elafibranor group. It was tolerated and safe, even though it caused a mild, reversible increase in serum creatinine [44]. The phase 3 RESOLVE-IT trial (NCT02704403) began in 2016 with the goal of recruiting 200 patients. Active recruitment will be completed in Dec 2021 [40].
5. Pan-caspase inhibitor: emricasan
Caspases are key enzymes in the apoptotic pathway. The detailed mechanism of action is shown in Fig. 2. Emricasan is a pan-caspase inhibitor which blocks apoptosis. In phase 2 clinical trial (NCT02077374), 28-day emricasan therapy decreased ALT, cytokeratin 18, and caspase 3/7 significantly. It was safe and well-tolerated [45]. In the clinical study of 23 patients with compensated cirrhosis, 28-day emricasan treatment decreased portal pressure in a subgroup of patients with severe portal hypertension (hepatic venous pressure gradient ≥12 mmHg) [46].
6. SCD1 modulator: aramchol
Stearoyl-coenzyme A desaturase 1 (SCD1) is a key enzyme which converts saturated fatty acid (SFA) to monounsaturated fatty acid (MUFA). SCD1 expression results in MUFA formation. In contrast, its deficiency results in SFA accumulation. Over-accumulation of SFA may result in ER stress and apoptosis [2]. Aramchol, a conjugate of cholic acid and arachidonic acid, is an inhibitor of SCD1. In a phase 2 clinical trial (n=58, NCT01094158), 300 mg aramchol treatment for 3 months decreased liver fat content and increased adiponectin levels significantly. Aramchol was safe and tolerable at a 300 mg dose [47]. Further data are lacking.
7. Monoclonal LOXL2 antibody: simtuzumab (GS-6624)
The lysyl oxidase (LOX) family is composed of four isoforms, LOX and the LOXL1-4. Among them, LOXL2 is a key contributor to collagen crosslinking stabilization. Moreover, LOX2 promotes hepatic progenitor cells towards the cholangiocyte lineage, while suppressing their differentiation into hepatocytes (Fig. 5) [15]. Theoretically, blocking LOX2 activity attenuates collagen crosslinking and fibrosis, and promotes liver regeneration. Based on this background, a phase 2b clinical trial of simtuzumab, an anti-LOXL2 monoclonal antibody, was conducted. Unfortunately, simtuzumab was ineffective in decreasing collagen content or the hepatic venous pressure gradient (NCT01672879) [16].
8. Fibroblast growth factor 19 agonist; NGM282
NGM282 is a variant of FGF-19, which reduces steatosis and lipotoxicity. In a phase 2 trial of 82 biopsy-proven NASH patients, 79% of the treatment group achieved the primary endpoint (≥5% reduction in absolute liver fat content by magnetic resonance imaging-proton density fat fraction after 12 weeks of treatment) compared to 7% in the placebo group (NCT02443116). ALT level decreased and LDL cholesterol was increased in the treatment group [48].
9. FGF-21 pegylated analogue; BMS-986036
BMS-986036 (Pegbelfermin) is a pegylated analogue of FGF-21. BMS-986036 decreased hepatic steatosis, NAFLD activity score (NAS) and fibrosis in a mouse NASH model, and improved insulin sensitivity, lipid profiles and fibrotic markers in obese diabetic patients. In a phase 2a trial of 75 obese biopsy-proven NASH patients, pegbelfermin achieved the primary endpoint and was tolerated during 16 weeks of treatment. The absolute hepatic fat fraction decreased 6.8% in the group who received a daily injection of 10 mg pegbelfermin, 5.2% in the 20mg weekly injection group, and 1.3% in the placebo injection group (p=0.0004, 0.008, respectively). Pegbelfermin had beneficial effects on adiponectin levels, lipid profiles, aminotransferase levels, serum pro-C3, and liver stiffness [49].
Conclusion
The pathophysiology of NASH progression is complex. Therefore, one drug targeting a single pathway may not be effective. That is the reason why many drugs failed in clinical trials. Although several drugs succeeded in phase 2 trials and moved on to phase 3, the efficacies were modest. Therefore, further research may be focused on combined therapy with two or more drugs covering different mechanisms of action.
 XML Download
XML Download