

 PDF
PDF Citation
Citation Print
Print



Introduction
The results of kidney transplantation (KT) have improved dramatically after the use of calcineurin inhibitors (CNIs) [1]. However, some complications of CNIs have been also reported [2]. Nephrotoxicity was a majorly reported side effect of CNIs that were assessed through laboratory or allograft biopsy findings [3,4]; however, it could be mitigated by controlling CNI trough levels. Unfortunately, mild neurologic symptoms of CNI neurotoxicity, such as tremors, agitation, insomnia, anxiety, and paresthesia, could easily go unnoticed [5,6]. In such cases, mild symptoms could worsen to cortical blindness, seizures, and encephalopathy, which could cause lethal damage to the brain. Recently, CNI neurotoxicity was diagnosed in computed tomography and magnetic resonance imaging studies, which showed morphological findings such as hypodensity of the white matter, cerebral edema, metabolic encephalopathy, and hypoxic damages [7-9]. Paradoxically, cyclosporine has been found to protect the brain from ischemia–reperfusion injury in animal models; however, the mechanism of CNI neurotoxicity is not yet fully understood.
Here, we investigated the neurotoxicity of two CNIs, cyclosporine, and tacrolimus, on the viability of glioma cells.
Materials and methods
1. Cell culture
Rat glioma cells (Korean Cell Line Bank, Seoul, Korea) were cultured in Dulbecco’s modified Eagle’s medium (WelGENE, Daegu, Korea) supplemented with 10% fetal bovine serum (WelGENE) and 1% penicillin–streptomycin (HyClone, Logan, UT, USA). Cells were grown in 10-cm diameter culture plates at 37℃ under humidified conditions containing 5% CO2/95% air.
2. Measuring the viability of glioma cells
Cytotoxicity was estimated by the 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay, which is based on the cleavage of a tetrazolium salt by mitochondrial dehydrogenases in viable cells. Glioma cells were plated in a 96-well plate at an initial density of 104 cells/well. After 24 hours, the medium was replaced with fresh medium containing various concentrations (0, 0.25, 0.5, 2.5, 5.0, and 10.0 mM) of cyclosporine (Novartis Pharma AG, Basel, Switzerland) or tacrolimus (Fujisawa Pharmaceutical Co., Ltd., Osaka, Japan). The drugs were dissolved in dimethyl sulfoxide, and in the experiment, the cells were diluted in DMEM medium and treated with the required concentrations. Cells were incubated at 37℃ for 24 hours. During the last 4 hours, the cells were incubated with 20 μL of MTT stock solution (5 mg/mL). The plates were shaken for 10 to 15 minutes in the dark. The optical density was measured at 570 nm using the microplate reader 550 (Bio-Rad, Lab., Hercules, CA, USA), and the relative cell viability was expressed using the following equation:
3. Statistical analysis
The values are expressed as mean±standard deviation. Statistical evaluation of the significant difference between the means was performed using Student’s t-test. SPSS version 18.0 (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses. p<0.05 was considered to be significant.
Results
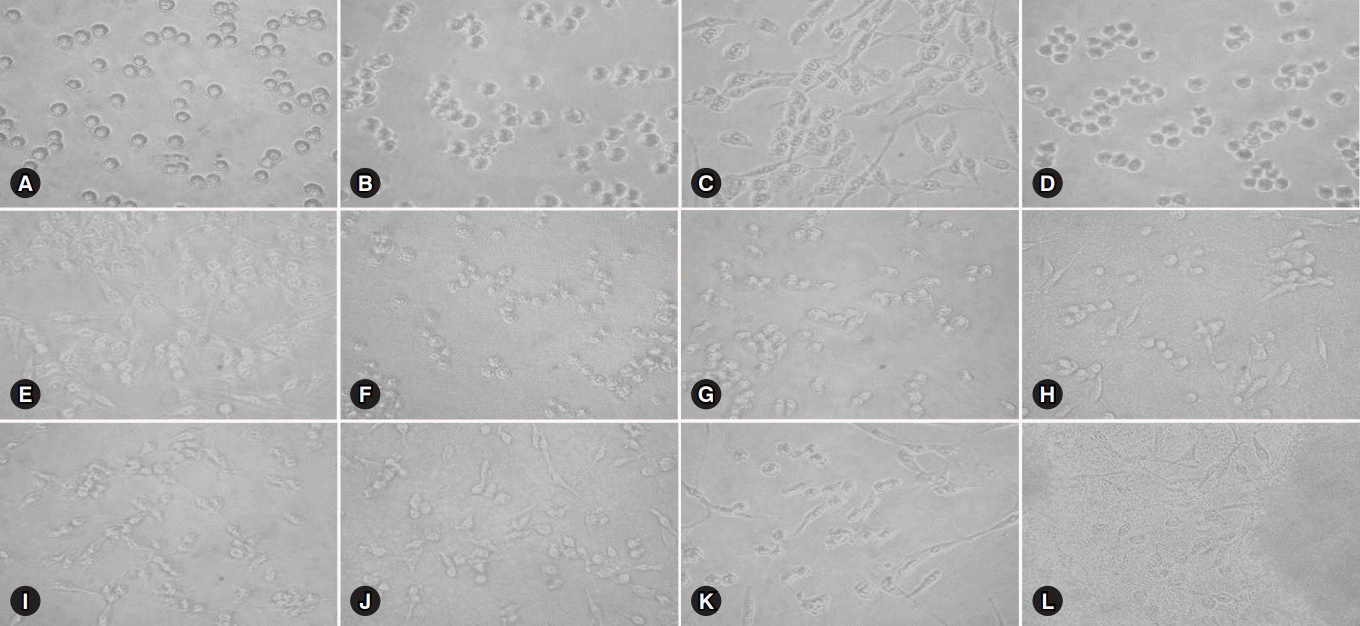
Substantial morphological changes were observed in glioma cells when they were treated with cyclosporine or tacrolimus. The cells were observed at a magnification of ×400 using the DMi1 inverted microscope (Leica Microsystems, Wetzlar, Germany). Cells were detached and floated to the top of the culture dish; however, a monolayer was not formed (Fig. 1).
For cyclosporine, cell viabilities were as follows: 100±0.1% at 0 mM/L cyclosporine control, 64.3±18.5% (p<0.05 vs. control) at 0.25 mM/L cyclosporine, 61.3±12.0% (p<0.01 vs. control) at 0.50 mM/L cyclosporine, 68.1±18.8% (p<0.05 vs. control) at 2.5 mM/L cyclosporine, 62.4±24.5% (p<0.05 vs. control) at 5.0 mM/L cyclosporine, and 68.6±19.5% (p<0.05 vs. control) at 10.0 mM/L cyclosporine.
For tacrolimus, cell viabilities were as follows: 100±0.1% at 0 mM/L tacrolimus control, 38.6±29.4% (p<0.05 vs. control) at 0.25 mM/L tacrolimus, 40.8±26.5% (p<0.05 vs. control) at 0.50 mM/L tacrolimus, 43.7±21.7% (p<0.05 vs. control) at 2.5 mM/L tacrolimus, 37.8±27.7% (p<0.01 vs. control) at 5.0 mM/L tacrolimus, and 43.0±29.8% (p<0.05 vs. control) at 10.0 mM/L tacrolimus.
Our study showed that the direct toxic effect of tacrolimus was stronger on brain cells than that of cyclosporine at the same concentration; however, no significant difference was observed between the two groups.
Discussion
Although advances in immunosuppressants have resulted in remarkable improvements with respect to allograft acceptance and patient survival rates in KT [1], the use of CNIs, such as cyclosporine and tacrolimus, has caused several side effects including nephrotoxicity [4], post-transplant bone disease [10], hepatotoxicity [11], hypertension [12], diabetes [13], dyslipidemia [2], and neurological side effects [14,15].
The clinical characteristics and mechanisms of CNI neurotoxicity are still controversial and poorly under¬stood. Recently, hypomagnesemia [16], hypocholesterolemia [17], severe vasoconstriction [17], and hypertension via the inhibition of nitric oxide production were reported as precipitating factors [18]; however, they do not sufficiently explain the mechanism of CNI neurotoxicity. In our previous study, cyclosporine and tacrolimus were shown to exhibit cytotoxic effects on renal cells and osteoblasts [10,19,20]. Thus, cyclosporine and tacrolimus may also exhibit cytotoxic effects on brain cells. Here, we studied the direct cytotoxicity of cyclosporine and tacrolimus on glioma cell viability to understand the underlying mechanisms of neurotoxicity.
Our results demonstrated that treatment with cyclosporine and tacrolimus resulted in substantial morphological changes as well as cell death in glioma cells.
Although neurotoxicity is known to be frequently associated with CNI trough levels exceeding recommended levels, it also occurs during long-term treatment even when CNI concentrations are within the therapeutic target range. Toxicity may be also be related to the type and dose of CNIs. For instance, tacrolimus is well known to be more potent than cyclosporine. In our study, the direct toxic effect of tacrolimus on glioma cell viability was greater than that of cyclosporine at the same concentration; however, no significant difference was observed. The clinical predisposition and mechanisms of cyclosporine-induced neurotoxicity remain controversial and poorly understood. Further studies are needed to investigate the precipitating factors for CNI-induced central nervous system abnormalities in addition to elucidating the positive effects of CNIs.
Interestingly, cyclosporine was previously reported to improve cerebral ischemia–reperfusion injury in vivo, and low concentrations of cyclosporine were shown to be neuroprotective [21]. Contrastingly, high concentrations of cyclosporine can cause mitochondrial dysfunction, which can lead to the deterioration of energy production, increased oxidative stress [22], and rapid apoptotic or necrotic cell death. Therefore, properly controlling CNI concentrations will be important in preventing CNI neurotoxicity.
CNIs can cause neurological side effects via direct cytotoxic effects in rat glioma cells. Therefore, we should carefully monitor neurologic symptoms and levels of CNIs.
 XML Download
XML Download