

 PDF
PDF Citation
Citation Print
Print



Introduction
Malignant peripheral nerve sheath tumors (MPNSTs) are rare, accounting for 5–10% of soft tissue sarcomas [1-3]. Half of MPNSTs occur in patients with neurofibromatosis type I (NF1), and the remainder are mostly sporadic MPNSTs (sMPNSTs). NF-MPNSTs have aggressive clinical features and a poor prognosis; after radical excision, NF-MPNSTs have high rates of recurrence rate and metastasis [2]. Most MPNSTs are single lesions; distant metastasis to skin tissue, soft tissue, or lymph nodes is rarely reported [3]. The most common site of lesion is the extremities, nerve root, or pelvic plexus [2], although MPNSTs can arise at any site of the human body [2]. Magnetic resolution imaging is very useful for diagnosing MPNSTs, but most MPNSTs are diagnosed with histologic testing after excision. Superficial MPNST in the cutaneous or subcutaneous tissue is very rarely found without NF1. Moreover, recurrent neurofibroma that transforms to MPNST is extremely rare, with only three cases reported by Chen et al. [4].
Case
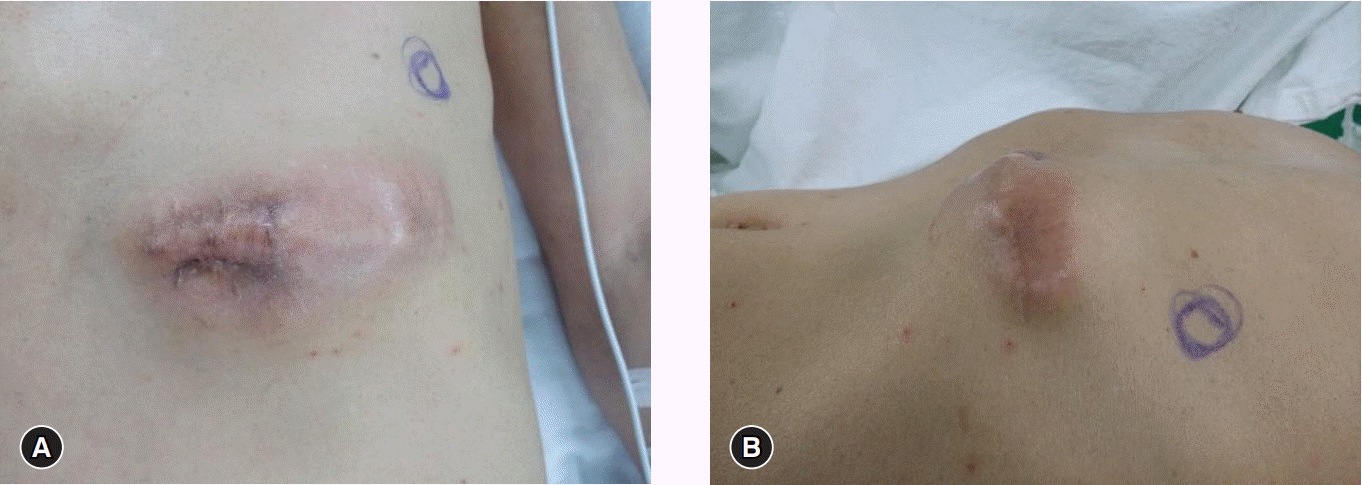
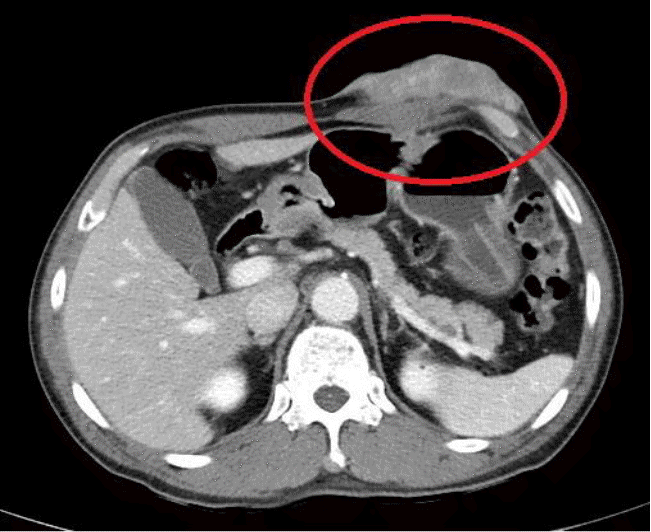
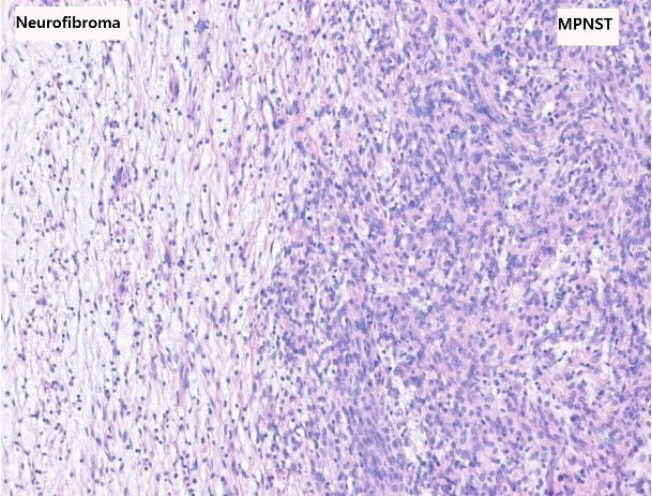
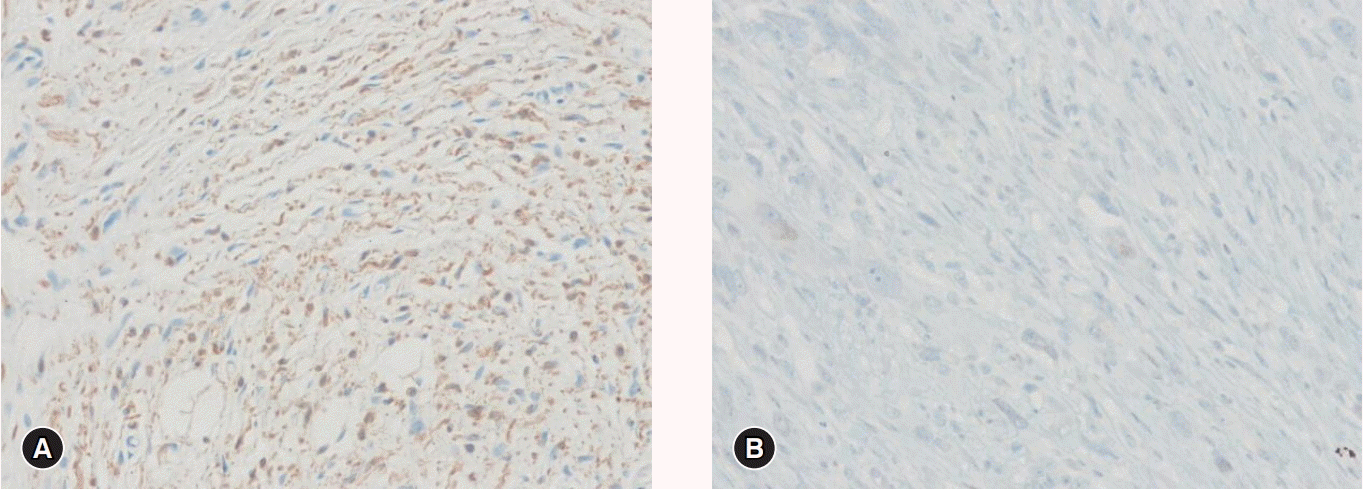
A 62-year-old man attended our hospital because of a recurrent neurofibroma in the left upper quadrant (LUQ) area of the abdominal subcutaneous tissue since 2009. With the exception of a prior myocardial infarction, he had not had other diseases. Since 2009, he had had excision and biopsy of neurofibroma in the subcutaneous tissue of the LUQ area of the abdomen three times; after the fourth, the patient had recurrence so he was referred to our hospital. On physical examination, there were no specific lesions, except an old operation scar and palpable mass in the LUQ of the abdomen (Fig. 1). Laboratory tests are non-specific. On computed tomography (CT), a 5×6-cm tumor was observed between the skin and subcutaneous tissue in the LLQ of the abdomen (Fig. 2). Under suspicion of neurofibroma, the patient was admitted and scheduled for operation. Under general anesthesia, the lesion was radically excised, and a flap and skin graft were done. The flap failed 17 days after the first operation, so a local advanced flap and skin graft were performed. Nineteen days after the second operation, the patient was discharged. Histologic testing showed tissue with specific spindle cells that were longitudinal and wavy, transformed from neurofibroma with low cellularity to MPNST with increased cellularity and pleomorphic cells (Fig. 3). On immunochemistry testing, an S100 protein test was weakly positive, and H3K27me3 staining was negative (Fig. 4); based on these results, we made a definite diagnosis of MPNST. The patient was regularly followed up in the departments of hemato-oncology and radio-oncology and treated with radiation therapy; he had no recurrence at 3 months.
Discussion
MPNSTs were previously named malignant schwannomas, for Schwann cells. MPNSTs are known to originate from a complex including Schwann cells, fibroblasts, and perineural cells. In 1978, malignant schwannomas, malignant nerve sheath tumors, neurosarcoma, and neurofibrosarcomas were grouped together as MPNSTs by the World Health Organization [5]. MPNST is a malignant soft tissue sarcoma originating from peripheral nerves or soft tissues, and they can arise in any site of the body [2,3,6,7]. MPNSTs comprise 5–10% of all sarcomas [1-3], and they occur in 1 person per million per year. In addition, MPNST of the skin or subcutaneous layer is very rare [8]. NF1 MPNSTs account for about half of all MPNSTs; the remainder are nearly all sMPNSTs [1,4]. The median age of patients with sMPNSTs is between 30 and 60 years [2]; the size of these tumors is smaller, and the stage shows less progression than NF-MPNSTs [1]. In the present case, the patient was 62 years old, close to the median age range for sMPNSTs. This was a very rare case of superficial sMPNST from recurrent neurofibroma in the subcutaneous layer. In 2015, Feng et al. investigated 13 patients admitted to Taipei Veterans General Hospital from 1999 to 2014 with superficial MPNST; of these, only three patients had body lesions [9]. Thomas et al. reviewed 230 cases of malignant spindle cell lesions of the skin, and only 2% of lesions were cutaneous MPNST [10]. However, there are no official statistics regarding MPNST occurrence, and most published articles are case reports.
Neurofibroma is a benign lesion that originates on the peripheral nerve sheath, the same as MPNST [11]. The most common sites of neurofibromas are cutaneous nerves of the body, neck, and head. A single lesion of neurofibroma has no specific symptoms [11]. The treatment of choice is excision; the prognosis of neurofibroma is better than that of MPNST, and postoperative malignant changes or local recurrences are very rare [11]. However, our patient showed both local recurrence and a malignant change.
Specific histologic features are very important in the diagnosis of MPNST. Neurofibroma, atypical neurofibroma, and MPNST are continuous progression lesions, making it difficult to distinguish them [7]. However, three features can be used to distinguish MPNST from neurofibromas: nuclear atypia, hypercellularity, and mitotic activity [7]. Both neurofibromas and MPNSTs comprise spindle cells, but neurofibromas have more collagen fibers than MPNSTs, which serves as another point of distinction between them [12,13]. In the present case, there was a specific transformation zone from the neurofibroma to the MPNST that showed increased cellularity and nuclear atypia.
Despite advancements in immunochemistry and molecular biology of sarcoma, diagnosis of MPNST remains difficult [13]. Recently, S100 protein has been reported as an important single marker for diagnosing MPNST. S100 protein has a nonspecific expression pattern but it has strong reproducibility [5]. However, there is a limitation that S100 protein is often negative in high-grade MPNST [14]. S100 protein is weakly positive in 50% of MPNSTs, but it is diffuse and shows a strong positive reaction in neurofibromas. Therefore, when S100 protein is diffuse and demonstrates a strong reaction, we can exclude possible MPNST [2,7]. Due to PRC2 inactivation in 70–90% of MPNSTs, tumor differentiation and progression is promoted, and demethylation of H3K27me3 (histone H3 lysine 27 trimethylation) develops [9]. Some researchers have highlighted demethylation of H3K27me3 in diagnosing MPNST with negative expression of H3K27me3. In about 51% of MPNSTs, H3K27me3 shows negative expression [9]. Studies of SOX10, EGFR, p53, and other markers are ongoing [15, 16], but there is no single marker that can be used for a definite diagnosis MPNST. In this case, expression of S100 protein was weakly positive and H3K27me3 was negative, so these were helpful in diagnosing MPNST.
This patient had had surgery three times, and biopsy results had repeatedly revealed neurofibroma. We initially believed he might have neurofibroma, but in a fourth excision and biopsy, we determined that his diagnosis was superficial MPNST from recurrent neurofibroma. Both neurofibroma and MPNST are rare diseases. Gross morphologies and images of both diseases are difficult to distinguish, and both have similar histologic features, so differential diagnosis of these diseases is very difficult. Because MPNST has poor prognosis, it is necessary to distinguish these pathologies. Especially for superficial MPNSTs in the cutaneous or subcutaneous layer, it is easier to find lesions than for deep MPNSTs. Early detection and treatment will lead to better prognosis [8]. Generally, superficial tumors in the cutaneous or subcutaneous layers are easy to see in clinical environments. For more precise diagnosis and treatment, excision is necessary, to yield a negative specimen margin. In patients with recurrent tumor, malignant changes should be considered, and after excisional surgeries, physicians should perform additional testing, such as immunochemistry and so on.
 XML Download
XML Download