

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Fabry disease (FD) is an X-linked, recessively inherited, rare, progressive, disorder of glycosphingolipid metabolism affecting multiple organs and resulting in organ dysfunction [1]. The incidence of FD is 1:117,000 live births and among them, very few “de novo” mutations have been detected [2,3]. The most frequently reported clinical manifestations of FD include renal failure, cardiomyopathy, and early cerebrovascular events. However, ocular lesions are often an early sign of the disease and can be detected in eye examinations [4]. One recent study showed that the most common ocular finding in FD is cornea verticillata, which is reported in up to 95% of male patients and 88% of female carriers [5]. Cataract was found in approximately 70% of male and 35% of females; tortuosity of conjunctival and retinal vessels was present in up to 77% of males and in approximately 20% of female patients. Extra-corneal lesions are almost always accompanied by cornea verticillata [4]. In Korea, there was a case of nine FD patients in one family presented with cornea verticillata. However there was no case report about the vessel abnormalities using various kinds of imaging modalities in de novo FD patient [6]. Here, the authors report a case of confirmed FD without a positive family history (de novo mutation), who underwent ophthalmologic examination and showed cornea verticillata and conjunctival vessel abnormalities.
Go to : 
CASE
A 41-year-old Asian male diagnosed with FD visited the clinic for ocular evaluation. The patient had been healthy until the age of 28 years, except for mild hypohidrosis since childhood. At the age of 28 years, the proteinuria was detected in regular medical checkup. He underwent renal biopsy and diagnosed with FD. There was no apparent family history of FD. Therefore, genetic analysis was performed to family members of the patient (mother and older brother) and none of them showed any alteration in the GLA gene, however, the patient had a mutation in hemizygosis at nucleotide c.293 C>T, suggesting a de novo mutation in patient (Fig. 1). He also had left ventricular hypertrophy and undergoing hemodialysis for chronic kidney disease. There was no history of systemic drug therapy that could develop vortex keratopathy. Slit lamp examination, fundus photography, fluorescein angiography, indocyanine green angiography (HRA-2; Heidelberg Engineering, Dossenheim, Germany), and optical coherence tomography (DRI OCT-1; Topcon, Tokyo, Japan) were performed. On examination, both eyes showed 20/20 best-corrected visual acuity and 14mmHg intraocular pressure. The slit lamp examination revealed a bilateral, paracentral, whorl-like pattern of white-to-brown corneal deposits (Figs. 2A, 2B). Tortuosity of episcleral vessels was noted in the right eye (Fig. 2C). Both eyes showed tortuosity and aneurysmal dilatation of the inferior bulbar conjunctival vessels (Figs. 2D, 2E). Cataract and retinal vessel tortuosity were not observed, and other imaging modalities demonstrated unremarkable findings (Figs. 2F, 3). As we considered the corneal and conjunctival abnormalites are the clinical signs of Fabry disease and the patient had been treated with enzyme replacement therapy (ERT), we decided to observe the ophthalmic findings.
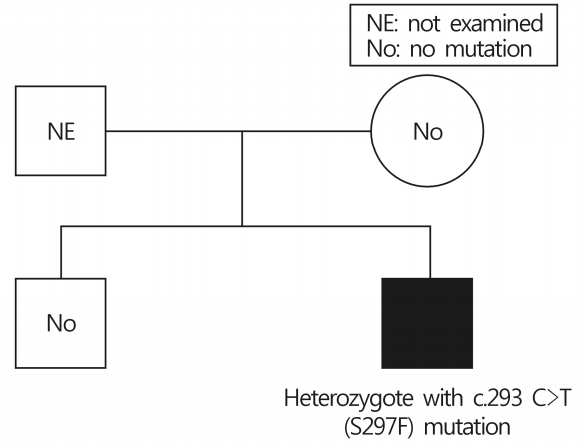 | Fig. 1.The family tree of de novo Fabry disease. The patient had a mutation in hemizygosis at nucleotide c.293 C>T, this mutation was not detected from his mother and older brother.
|
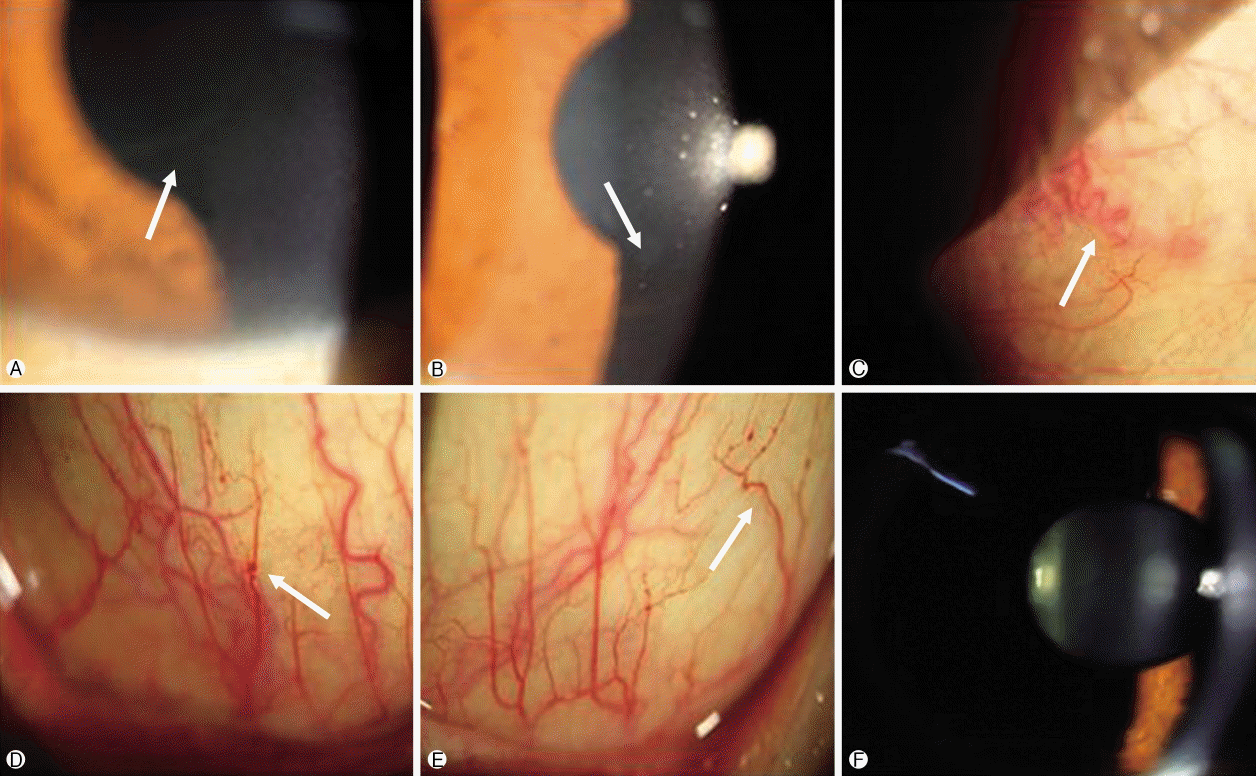 | Fig. 2.Photographs of a patient with de novo Fabry disease. Cornea verticillata (arrow) show whorl-like pattern of white, brown corneal opacity in the right eye (A) and left eye (B). (C) Tortuosity and dilatation of episcleral vessel (arrow) of the right eye. Tortuosity and aneurysmal dilatation of the inferior bulbar conjunctiva vessels (arrow) in the right eye (D) and left eye (E). (F) Lens evalua- tion show no signs of Fabry cataract.
|
Go to :
DISCUSSION
FD is a rare X-linked hereditary disease. Due to its rarity, FD diagnosis remains a challenge. Furthermore, the diagnosis is even more difficult when family history is absent [7]. Delayed diagnosis of FD is still common; there is on average, a gap of a decade or more, between the onset of symptoms and diagnosis of FD. However, the ocular manifestations begin in childhood [4]. In one retrospective analysis from the United States; 26% of FD patients with no family history of the disease were diagnosed by an ophthalmologist, suggests that ophthalmic physicians can play an important role in diagnosis of FD[8].
In this case, a detailed ocular examination was performed to evaluate this patient with de novo FD. We identified cornea verticillata and tortuous, aneurysmal dilatation of the inferior bulbar conjunctival vessels on slit lamp examination. These findings support that the extra-corneal lesions are accompanied by cornea verticillata and the vascular abnormalities are most often seen on the inferior bulbar conjunctiva. However, other abnormalities such as Fabry cataract and retinal vessel disorders were not observed. There have been reports that corneal and conjunctival vascular abnormalities manifest at a younger age than lens or retinal disorders in FD [4]. The patient is on ERT and this may have slowed down disease progression.
This case highlights the fact that ophthalmologists should always be aware of the possibility of de novo FD in patients with corneal verticillata and the inferior bulbar conjunctival vessels abnormalities, even in the absence of a family history.
In conclusion, we report the case of de novo FD patient who underwent comprehensive ophthalmological evaluations using various kinds of imaging modalities and showed cornea verticillata and inferior conjunctival vessel abnormalities.
Go to :
 XML Download
XML Download