

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
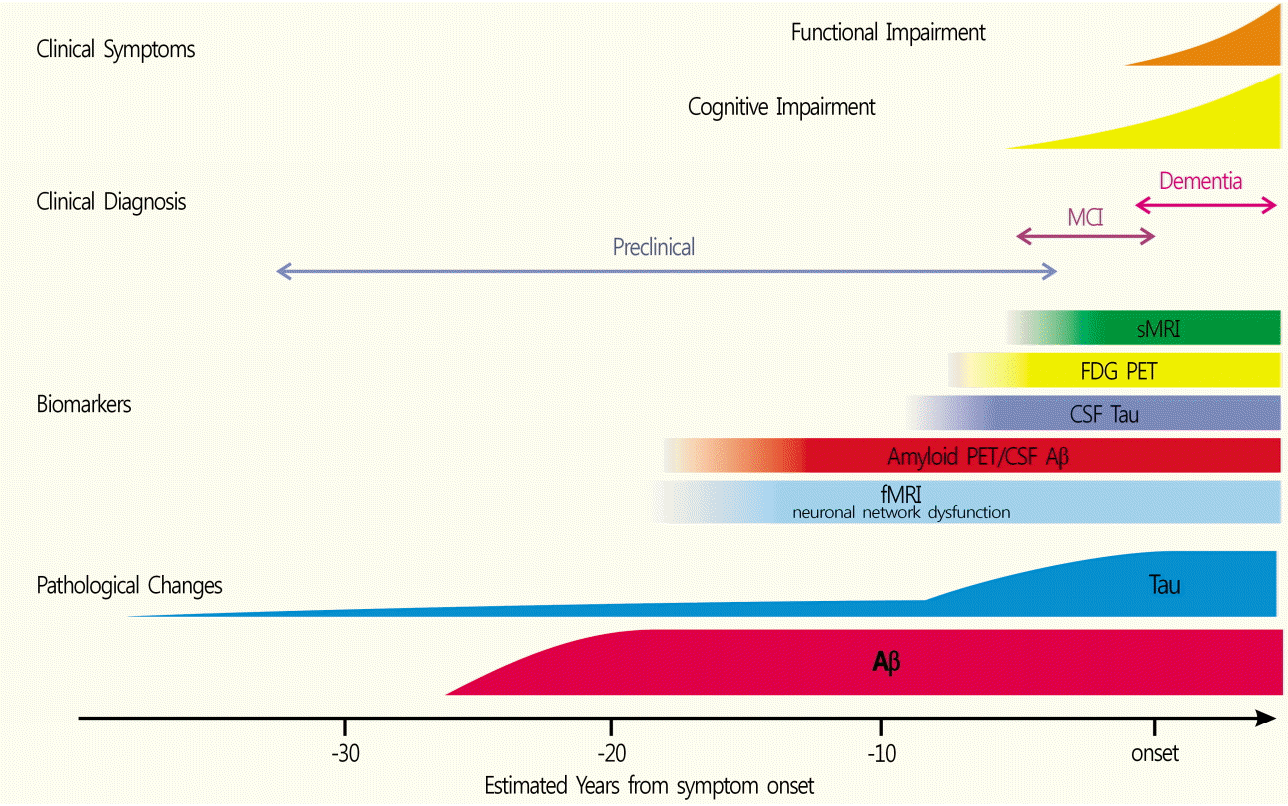
With the number of dementia patients rising due to an aging population, it is important to detect dementia early and delay its progression. A variety of diagnostic methods are being used for this purpose, including mental status examination, neurophysiological testing, and magnetic resonance imaging (MRI). Alzheimer’s disease (AD) is one of the most common types of dementia, accounting for 70% of all cases. It is pathologically characterized by deposition of beta-amyloid (Aβ), forming amyloid plaques outside neurons, and an accumulation of hyperphosphorylated tau proteins inside neurons, both of which are known to induce neuronal apoptosis and ultimately cause dementia [1]. Abnormal deposition of Aβ is subsequently followed by synaptic dysfunction, tau protein abnormality in the cerebrospinal fluid (CSF), structural changes of the brain, and cognitive decline (Fig. 1) [2]. Accumulation of Aβ starts at an early stage of the disease, when no symptoms are present. Studies investigating early biomarkers to differentiate AD and normal individuals have attempted to predict the progression or onset of AD from mild cognitive impairment —the intermediate stage between healthy state and dementia —or the preclinical stage —the stage preceding mild cognitive impairment —and halt the progression of the disease [3]. Accumulated Aβ can be observed in an autopsy or Aβ1-42 can be detected in the CSF; currently, the extent of Aβ accumulation can be visualized in living patients using neuroimaging. The methods used to visualize Aβ include positron emission tomography (PET), and new compounds have been developed and approved by the US Food and Drug Administration (FDA) since the development of the C-11 Pittsburgh compound B (PiB) in 2004. Although a fundamental treatment for AD is still lacking, efforts to develop new therapeutic agents are ongoing, which would serve as useful tests for the diagnosis performed using Aβ PET, and the assessment of treatment effectiveness [4].
Beta-amyloid
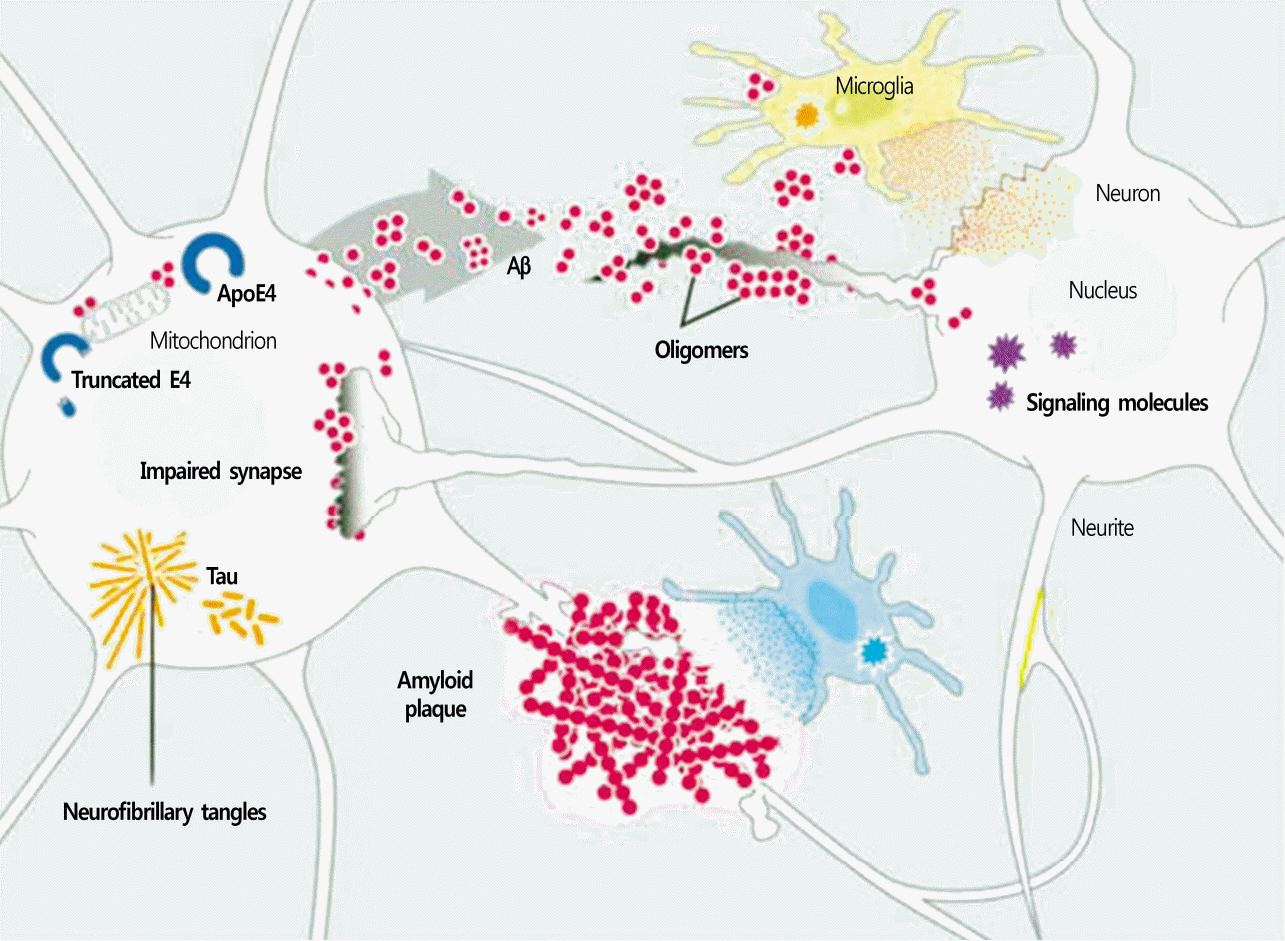
Found in the brain of patients with AD, Aβ is a peptide of 36-43 amino acids, and when it forms plaques, it becomes neurotoxic. This peptide is produced by the action of β-secretase and γ-secretase on the amyloid precursor protein [5]. Subsequently, Aβ is converted to amyloid fibrils via a nucleation reaction, after which it accumulates coagulates [6]. Amyloid plaques, which primarily consists of amyloid fibrils, are surrounded by axons, dendrites, reactive astrocytes, and activated microglia (Fig. 2), and Aβ is detected not only in amyloid plaques but also in cortical arteries, CSF, plasma, and neuronal culture [7]. Amyloid plaques can be classified according to density and structure into diffuse plaques, which have a low fibril content, and neuritic plaques, which have a high fibril content [8], with the latter type known to be the more significant indicator of AD[9].
Although the exact physiological role of Aβ has not yet been revealed, evidences suggest that it is important for the regulation of synaptic activities and survival of neurons [10]. All available genetic, pathological, biochemical, and cytological evidences support that the gradual accumulation of Aβ in the brain, as a result of an imbalance of the production and removal of Aβ, is an important factor in the etiology of AD, and despite conflicting arguments, Aβ is still an important risk factor for AD[11,12].
Beta-amyloid imaging
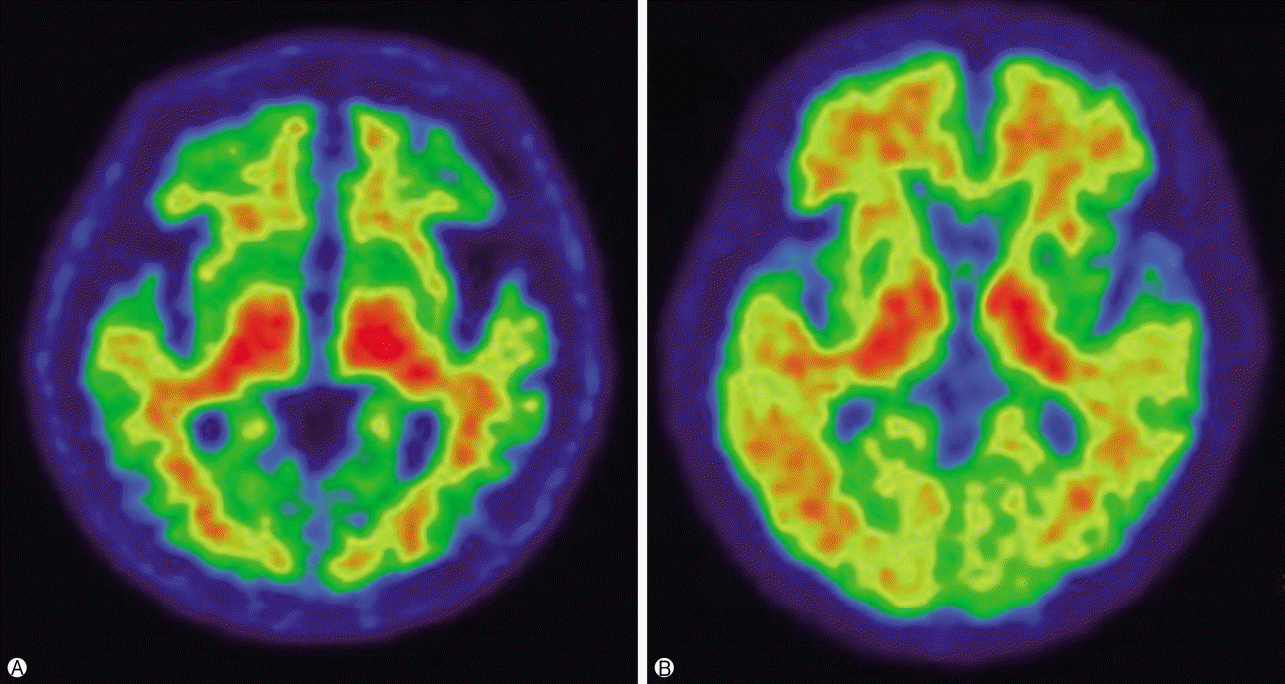
Radiopharmaceuticals for Aβ imaging have been developed based on the chemical structures of the dyes used to stain Aβ in the conventional pathologic examination. These substances were modified by eliminating their ionization properties, and enhancing their lipophilic properties, such that they could penetrate the blood brain barrier, and accumulate in the parts of the brain containing Aβ aggregates [13]. Most Aβ imaging methods involve the use of a PET tracer known as 11C-PiB, but its use is limited due to its short half-life (20 min). Recently, a fluoride (18F) marker with a long half-life (110 min) has been used in several ongoing clinical trials; of them, Amyvid (florbetapir; 18F-AV-45) was the first to be approved by the FDA in 2012, followed by Vizamyl (flutemetamol; GE-067) in 2013, and Neuraceq (florbetaben; BAY 94-9172) in 2014. Currently, Neuraceq and Vizamyl are being used in South Korea. Owing to its high sensitivity, Aβ imaging enables early diagnosis, and can predict the progression of mild cognitive impairment to AD. In particular, a two-year follow-up study of patients with mild cognitive impairment reported that those who progressed to AD had a higher uptake of 11C-PiB than those who did not convert to AD, suggesting that Aβ PET is useful for the early diagnosis of AD [13]. In a study that examined the local deposition of Aβ radiological tracers, and the presence of local amyloid plaques reported in autopsies or biopsies of patients with AD, the agreement rate between the two exceeded 96%[14,15]. The deposition of Aβ is usually observed in the cerebral cortex of patients with AD, that is, it is deposited in the gray matter, including the prefrontal, medial parietal, lateral temporal, and cingulate gyrus and subcortical striatum(Fig. 3) [16,17]. Images are analyzed and determined as positive or negative by comparing the radioactivity between the grey matter in the cerebral cortex and the proximal white matter. When negative, the contrast between the white and grey matters is clear, showing elevated radioactivity in the white matter as opposed to the grey matter; however, when positive, the contrast between the two is weakened, and it is difficult to distinguish the white matter from the grey matter. Therefore, Aβ PET images can provide additional information to confirm the diagnosis in cases where the diagnosis of AD is uncertain, and are also highly useful for distinguishing between frontotemporal dementia and AD [18]. However, such Aβ images are not specific to AD, as they are positive also for dementia with Lewy bodies and cerebral amyloid angiopathy, hindering differential diagnosis [19]. Therefore, Aβ images should be accepted as a general marker of cerebral amyloidosis, not a specific marker for amyloidosis in the patients with AD.
Moreover, Aβ images can be meaningful even when observed in dementia other than AD. In dementia with Lewy body, high Aβ deposition is associated with a more severe cognitive impairment, and a better response to choline esterase inhibitors, while Aβ deposition in Parkinson’s disease signifies rapid progression to dementia [20-22]. These findings suggest that α synucleinopathy can coexist with the pathology of AD[23]. However, one shortcoming is that the possibility of testing positive for Aβ increases with age, and amyloid plaques can be observed on Aβ PET even in elderly individuals with normal cognitive function. In fact, positive findings can be observed in under 5, 10, 25, and 50% of the people in their 50s-60s, 60s-70s, 70s-80s, and 80s-90s, respectively [24,25]. Even in patients without dementia, the prevalence of cerebral Aβ pathology, determined based on Aβ PET or CSF findings, is associated with age, and the formation of amyloid plaques in patients with dementia can begin up to 20 years prior to the onset of clinical symptoms [26,27]. Thus, indications for Aβ PET application have been suggested as follows: first, patients with persistent or progressive mild cognitive impairment with an unknown cause; second, patients with a possibility of AD, with unclear clinical symptoms, and third, relatively younger patients (aged 65 years or less) with progressive dementia. It is generally not performed for determining the severity of symptoms [28].
In addition to Aβ PET, computed tomography or brain MRI are also used to measure brain volume, shape, and intensity to assess the loss or atrophy of brain tissue to identify the pathophysiology of AD. Many studies have reported that atrophy of the medial temporal area is observed from an early stage of AD, and a meta-analysis of a brain volume study has reported a sensitivity of 78-94% and specificity of 60-100% for differentiating AD from normal condition [3,29]. In addition, changes in brain activities can be measured by regional blood flow or measuring glucose metabolism using fluorodeoxyglucose (FDG) PET. Studies using FDG-PET have found that metabolism is reduced in the medial temporal area, parietal area, and posterior cingulate gyrus. Reduced metabolism is also observed in the frontal lobe in more advance stages of AD. These patterns are in contrast with the preservation of the primary motor/visual cortex, cerebellum, thalamus, and basal ganglia [3,30]. An Aβ PET study using PiB differentiated between frontotemporal dementia and AD with an accuracy of 97%[18], while it differentiated between AD and other neurodegenerative diseases with an accuracy of 87.5% [31,32]. Interestingly, Aβ PET yields negative results for frontotemporal dementia, which helps in the differentiation of AD, and development of treatment plans. A recent study investigating the usefulness of Aβ images suggested that they lead to a change in diagnosis for about 30% of the patients, and a change in treatment policies for about 60% of the patients [33].
Although there is no cure for AD, symptomatic treatment using acetylcholinesterase inhibitors or glutamatergic modulators can bring about temporary stabilization, though not a marked improvement in memory [34]. Other treatment methods are currently being developed, one of which is antibody-based immunotherapy, which targets Aβ to inhibit the accumulation of or remove Aβ in the brain, delaying the progression of the disease. In a phase two clinical trial for bapineuzumab (a humanized anti-amyloid-beta monoclonal anti-body), Aβ PET images and tau protein concentrations in the CSF differed according to the presence of apolipoprotein E ε4, but the agent did not improve cognitive functions or other symptoms in patients with AD [35]. A recent study of aducanumab found that treatment led to reduction of amyloid plaques in Aβ PET images, and that the agent delayed the deterioration of cognition [36]. Therefore, Aβ PET imaging is expected to be useful in monitoring the progression of a disease or treatment effects by accurate diagnosis of AD in the early or preclinical stages, and measuring the Aβ accumulation in the brain [13].
CONCLUSION
The number of patients with dementia is rising due to an aging population, and most of these patients are diagnosed with AD. The disease is pathologically characterized by amyloid plaques outside neurons and accumulation of tau protein within neurons, which are known to induce AD by promoting neuronal apoptosis [1]. Detecting accumulated Aβ before the onset of symptoms is highly important, as individuals in this stage can potentially benefit from treatments that aim to reduce or remove Aβ in the brain before an irreversible loss of neurons or synapses occurs [37]. In this context, the importance of early diagnostic methods using imaging biomarkers has been emphasized, and PET scans that visualize Aβ are expected to play an important role in the early diagnosis of AD and the development of therapeutic agents [23].
 XML Download
XML Download