

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hepatoblastoma is the most common pediatric primary liver tumor and constitutes 90% of tumors in children aged 5 years or younger.1 During the last 30 years, the overall incidence of hepatoblastoma exhibited the greatest increase among all pediatric malignancies worldwide, which were related to increases in premature births, low birth weight and survival, as well as earlier detection and better imaging modalities. 2–4 Most cases are sporadic and occur in the liver without any background liver disease, but they are sometimes associated with constitutional genetic abnormalities, malformations, and familial cancer syndromes such as Beckwith-Wiedemann syndrome and familial adenomatous polyposis.5,6 Hepatoblastoma is an embryonic tumor believed to arise from hepatocellular precursors that often recapitulate stages of liver development. These tumors rarely consist of only one cell type and usually present a combination of epithelial, mesenchymal, undifferentiated, and other histological components.7–9 Hepatocellular carcinoma is the second most common malignant liver tumor diagnosed in children and pediatric well-differentiated hepatocellular carcinoma is histologically similar to hepatoblastoma, fetal subtype. Pediatric well-differential hepatocellular carcinoma is differentiated from fetal hepatoblastom by the presence of thickened trabeculae, high nucleus:cytoplasm ratios, the absence of light and dark areas, and usually negative β-catenin nuclear. Since hepatoblastoma is rarely observed, even in specialized institutions, and no international consensus classification existed until recently, even general pathologists find it difficult to make a diagnosis.7 The International Pediatric Liver Tumors Consensus Classification for hepatoblastoma was therefore established based on the discussion on histopathology and classification of hepatoblastoma at the International Symposium on Pathology in March 2011. The 5th Edition of the World Health Organization Classification of Digestive System Tumors (2019) classified the histological patterns of hepatoblastoma in accordance with the International Pediatric Liver Tumors Consensus Classification (Table 1). In the present review, we summarize the recently described histopathological classification of hepatoblastoma and immunohistochemistry used for diagnosis (Table 2).7
Table 1
Histological classification of hepatoblastoma
![]()
Table 2
Immunohistochemistry for the diagnosis of hepatoblastoma
![]()
Go to : 
EPITHELIAL TYPE
Fetal hepatoblastoma consists of thin trabeculae or nests of small to medium-sized cells and immunohistochemical expression of glutamine synthetase and Hep par-1 is helpful for differentiating the other subtypes.
1. Fetal with low mitotic activity (well-differentiated) subtype
The well-differentiated fetal subtype consists of cells measuring 10–20 μm in diameter that grow either as one- or two-cell thick cords with round nuclei, finely stippled chromatin, well-delineated nuclear membranes, and inconspicuous nucleoli. The cytoplasm is either clear or finely granular and eosinophilic, reflecting variable amounts of glycogen and lipids (Fig. 1A). Well-differentiated hepatoblastoma has minimal mitotic activity (<2/10 HFPs). The patterns of finely granular positivity of glypican 3 immunohistochemistry can be used to differentiate well-differentiated fetal hepatoblastoma from other fetal subtypes (Fig. 1B). Extramedullary hematopoiesis consisting of clusters of erythroid precursors is often present.
 | Figure 1Hepatoblastoma, epithelial type, fetal subtype. (A) Fetal with low mitotic activity (well-differentiated) subtype (hematoxylin & eosin [H&E], ×400) and (B) fine glypican 3 expression (×400). (C) Fetal, mitotically active (crowded fetal) subtype, mitosis (arrows) (H&E, ×400) and (D) strongly coarse glypican 3 expression (×400). (E) Fetal, pleomorphic subtype (H&E, ×400) and (F) strongly coarse expression of glypican 3 (×400).
|
2. Fetal, mitotically active (crowded fetal) subtype
Crowded fetal hepatoblastoma has well-delineated plasma membranes, but more amphophilic cytoplasm due to decreased cytoplasmic glycogen and a proportionately higher nuclear/cytoplasmic (N/C) ratio that results in a crowded hypercellular appearance and increased mitotic activity (≥2/10 HFPs) (Fig. 1C). Crowded fetal hepatoblastoma is often adjacent to embryonal areas and small cell undifferentiated, and a transition between the two patterns often occurs. Glypican 3 immunohistochemistry shows strongly coarse expression (Fig. 1D).
3. Fetal, pleomorphic subtype
Pleomorphic fetal hepatoblastoma is an uncommon subtype that is usually seen after chemotherapy and in metastases following chemotherapy administration. Individual cells contain pleomorphic nuclei with the following characteristics: irregular shape, large size, and conspicuous nucleoli with variably coarse chromatin (Fig. 1E). In most cases, glypican 3 exhibits a strongly coarse positivity such as that observed in the crowded fetal subtype, but rarely, negativity (Fig. 1F).
4. Embryonal subtype
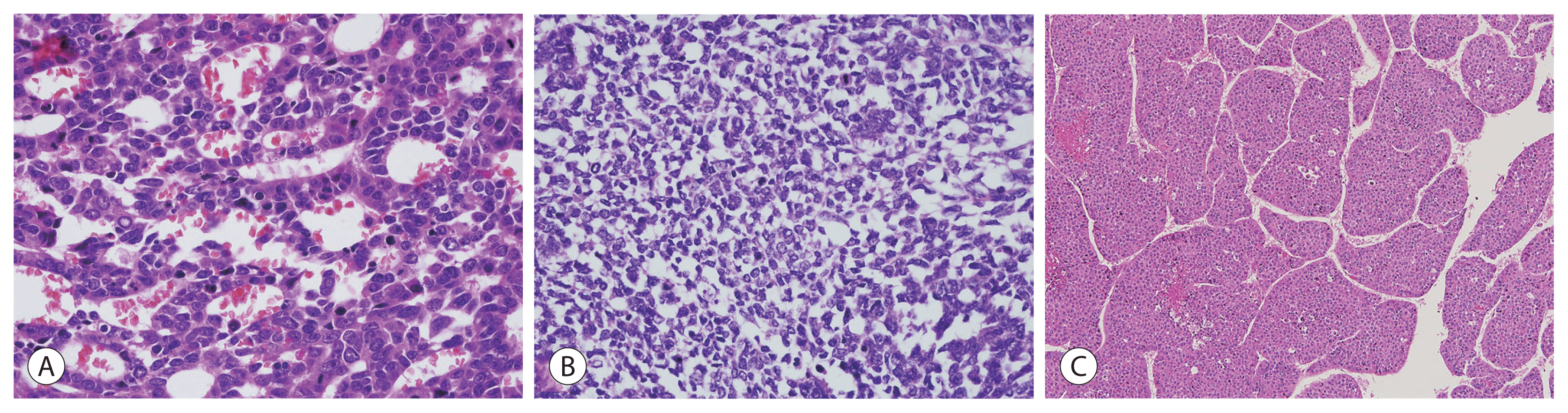
Embryonal hepatoblastoma resembles the developing liver at ~6–8 weeks of gestation and is arranged as solid nests or either glandular or acinar structures. Pseudorosettes and papillary configurations are also formed. The cells have scant, dark granular cytoplasm devoid of visible glycogen and lipid droplets and contain enlarged nuclei with coarse chromatin (Fig. 2A). Mitotic activity is higher in these cells than in the fetal type, and extramedullary hematopoiesis is rare.
5. Small-cell undifferentiated subtype
Small-cell undifferentiated (SCUD) hepatoblastoma is a clinically important subtype with low or normal serum alpha-fetoprotein (AFP) levels and is associated with an aggressive biology and worse survival rates. The tumor cells have a diffuse pattern, but usually form either nests or clusters intimately intermixed with epithelial cell types. Small cells are slightly larger than lymphocytes (usually 7–8 μm) and are round to oval-shaped with scant cytoplasm and relatively fine nuclear chromatin along with inconspicuous nucleoli (Fig. 2B). Apoptosis, necrosis, and mitotic figures are abundant. The immunohistochemical features of co-expression of pan-cytokeratin and vimentin indicate the SCUD subtype, even in histologically unclear cases, whereas the expression of AFP or glypican 3 does not. INI1-negative SCUD hepatoblastoma has features similar to those of rhabdoid tumors, which are characterized by discohesive, eccentric, irregular nuclei, prominent nucleoli, and abundant cytoplasmic filaments. Therefore, INI1-negative SCUD hepatoblastoma likely represents hepatic rhabdoid tumors. INI1-positive SCUD hepatoblastoma is associated with a better prognosis than INI1-negative SCUD hepatoblastoma.
6. Cholangioblastic subtype
Cholangioblastic hepatoblastoma can differentiate as cholangiocytes and form small ducts and express the cholangiocyte lineage markers cytokeratin 7 and 19. These components may be found within or around the hepatocellular component. The tumor cells of cholangioblastic hepatoblastoma are usually negative for glypican 3, which helps to differentiate them from embryonal hepatoblastoma, which is histologically similar. In addition, nuclear expression of beta-catenin in cholanigoblastic hepatoblastoma can be useful when differentiating reactive ductal proliferation from membranous expression of beta-catenin following chemotherapy.
7. Macrotrabecular subtype
Macrotrabecular hepatoblastoma accounts for <5% of all cases and is characterized by a thick cell plate of ~5–12-cell trabeculae. The trabeculae may consist of fetal, embryonal, or pleomorphic cells or cells that resemble those present in hepatocellular carcinoma (Fig. 2C).
Go to :
MIXED EPITHELIAL AND MESENCHYMAL TYPE
This classification designation comprises both epithelial and mesenchymal components.
1. Without teratoid feature subtype
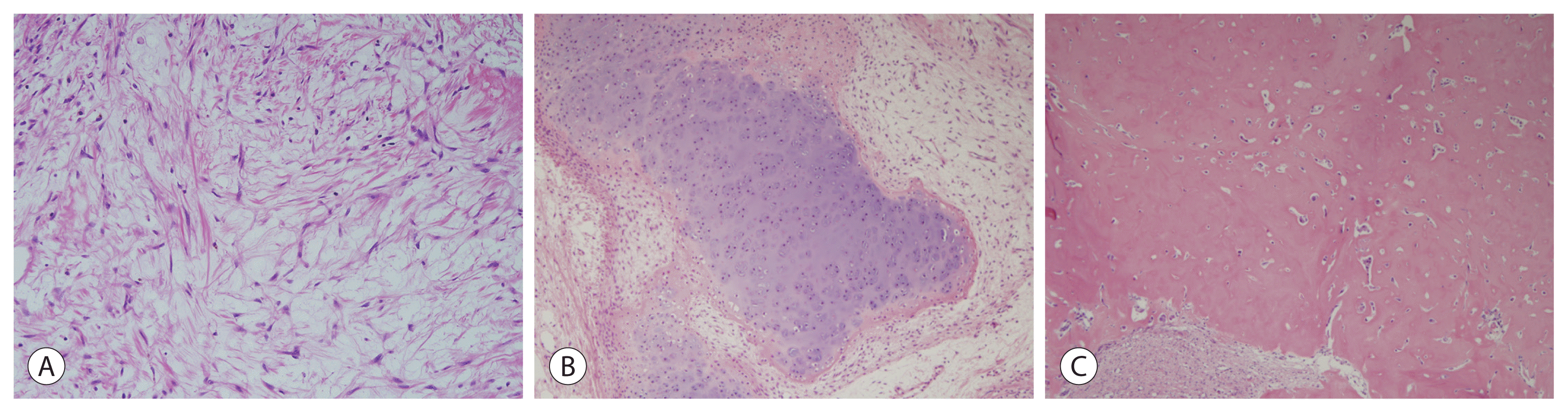
The mesenchymal component in mixed epithelial and mesenchymal hepatoblastoma comprises stromal derivatives with mature and immature fibrous tissue, osteoid or osteoid-like tissue, and hyaline cartilage (Fig. 3). The mesenchymal component represents an integral part of tumors and is distinct from chemotherapy-induced or metaplastic changes.
2. With teratoid features subtype
A small percentage of mixed epithelial and mesenchymal hepatoblastoma cases may exhibit stromal derivatives with teratoid features of neural or neuroectoderamal differentiation, which is represented by the mature brain, primitive neuroepithelial components forming tubules and rosettes, melanin, and retinal pigment. Squamous epithelium, mucinous glands, striated muscle, cartilage, and bone may also be present (Fig. 4).
 | Figure 4Hepatoblastoma, mixed epithelial and mesenchymal type with teratoid feature subtype. (A) Bone (H&E, ×100), (B) glial element (H&E, ×200), (C) melanin pigment (H&E, ×200), and (D) melanin pigment (Fontana-Masson stain, ×200). (E) Mucinous gland (H&E, ×200). (F) Mucinous gland (mucicarmine stain, ×200).
|
Go to :
CONCLUSION
Hepatoblastoma usually consists of a mixture of several components, including epithelial and mesenchymal cells, and each component has overlapping characteristics; therefore, distinguishing each subtype based on histological characteristics alone is difficult. The biopsy sample, which makes up <0.003% of the entire tumor, rarely represents the rest of the tumor.7 Therefore, histological analysis of remaining tumors after neoadjuvant chemotherapy is crucial. Immunohistochemisty helps to identify residual neoplastic cells and demarcate their boundaries in surgical specimens after chemotherapy. Immunohistochemistry may also assist in histological classification and risk stratification (Table 2).7 However, no marker to differentiate hepatoblastoma form hepatocellular carcinoma currently exists.
Go to :
 XML Download
XML Download