

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Primary ciliary dyskinesia (PCD) is a rare genetic disorder of the motile cilia/flagella characterized by situs inversus and male infertility. PCD leads to infancy-onset chronic respiratory diseases, including frequent respiratory infections and bronchiectasis [1]. Typically, PCD is diagnosed late, with a median age at diagnosis of 5.3 years, leading to poor lung condition and quality of life [2, 3]. We report a PCD case due to a novel exon deletion with the first Korean variant p.Gln833Valfs*6 in CCDC39 diagnosed at infancy via whole-genome sequencing (WGS). The Institutional Review Board of Jeonbuk National University Hospital (JNUH), Jeonju, Korea, approved this study (JBNUH 2022-01-039) and waived informed consent for this study.
In August 2020, A boy weighing 3,080 g was born at 38 weeks of gestational age in a local obstetrics clinic. A prenatal ultrasonography revealed dextrocardia. There was no familial history of congenital anomaly. Apgar scores at 1 and 5 minutes were 7 and 8 points, respectively. As subcostal retraction and tachypnea were observed, he was transferred to the Neonatal Intensive Care Unit in JNUH. Dextrocardia and situs inversus were confirmed on two-dimensional echocardiography and abdomen sonography, respectively. Based on situs inversus and unexplained respiratory difficulty in the neonatal period, we strongly suspected neonatal PCD [1]. As recommended by the American Thoracic Society clinical practice guideline for PCD, we decided to perform trio WGS to evaluate the genetic variants associated with PCD [4].
A sequencing library was prepared using the MGIEasy Universal DNA Library Prep Set kit (v1.0; MGI Tech Co., Ltd., Shenzhen, China) and was sequenced on the MGI DNBSEQ-T7 platform (MGI Tech), generating 2×100 bp paired-end reads. WGS revealed a heterozygous variant, c.2497_2498del (p.Gln833Valfs*6), in CCDC39, which is one of the causative genes of PCD (Fig. 1A). This variant is expected to create a premature translational stop signal and is classified as pathogenic in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). It has a minor allele frequency of 0.0015% in all ethnicities reported in gnomAD (https://gnomad.broadinstitute.org/); however, it is not reported in the Korean Reference Genome database (http://coda.nih.go.kr/coda/KRGDB/). Based on these pieces of evidence, we classified the variant as pathogenic according to the American College Medical Genetics and Genomics guidelines [5]. The variant was also detected in the patient’s father (Fig. 1B).
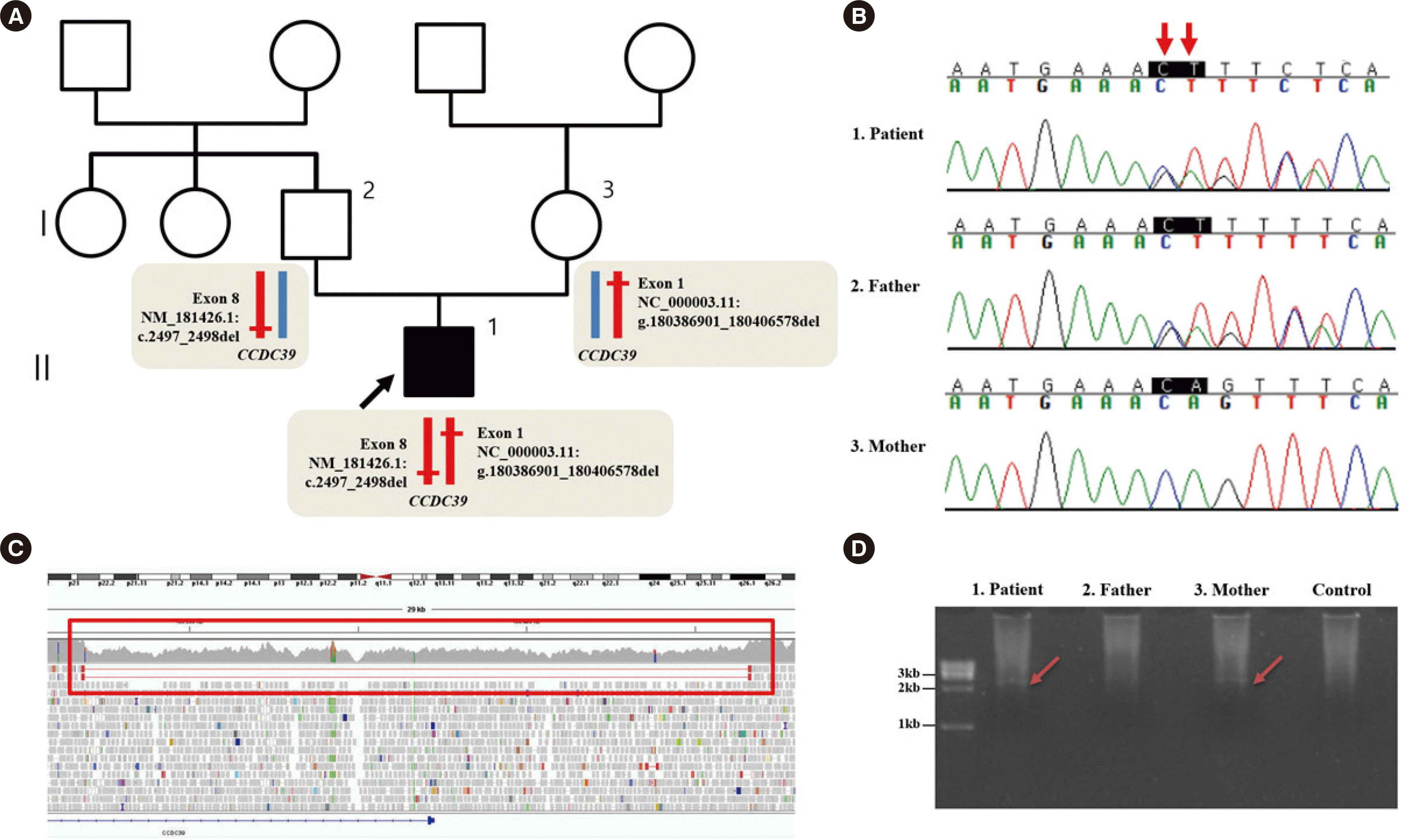 | Fig. 1Pedigree and genetic evaluation of the patient. (A) Pedigree with mutations indicated. The black arrow indicates the proband. The schematic alleles show the heterozygous variant in CCDC39. (B) Sanger sequencing traces for the patient and his father and mother. The variant c.2497_2498del (red arrow) in CCDC39 was confirmed to be inherited from the father (reference sequence: NM_181426.1). (C) The red rectangle indicates WGS data showing a lower sequencing depth (39.7X) than the adjacent area (71.5X) and paired-end reads with a deletion of the region 180,386,901–180,406,578 in chromosome 3 [hg19]. (D) Gap-PCR revealed a 2.7 kb-sized band corresponding to deletion of the 5’ UTR and exon 1 of CCDC39 in the patient and his mother.
Abbreviations: WGS, whole genome sequencing; UTR, untranslated region.
|
WGS also revealed paired-end reads with an insert size larger than expected. The inferred deletion size by CNVnator was 19,678bp (NC_000003.11:g.180386901_180406578del) (Fig. 1C). This genome region overlaps with the 5` UTR and exon 1 of CCDC39. This novel structural variant has not been reported in gnomAD SVs v2.1, Database of Genomic Variants Gold Standard dataset (http://dgv.tcag.ca/dgv/app/home), ClinVar, and Human Gene Mutation Database Professional (v2021.2; http://www.hgmd.cf.ac.uk/). The deletion was identified in the patient and his mother by gap-PCR (Fig. 1D). DNA was amplified in a 50-µL reaction mixture containing 5 µL of 10× PCR buffer, 5 µL of DMSO, 0.75 µL of Expand Long Template Enzyme Mix, and 7.5 pmol of each primer. The primers used to amplify the breakpoint region were 5´-ATGGCTCATGCCTGTAATCC-3´ and 5´-TCACTGATTCAACAAAAACTTCA-3´. The expected amplicon size was 2,724 bp. PCRs were run in an ABI 9700 Thermal Cycler (Life Technologies, Foster City, CA, USA) according to the manufacturer’s instructions (Roche, Mannheim, Germany). However, the breakpoints could not be confirmed by Sanger sequencing as they were located at repeat regions, such as long interspersed nuclear elements and segmental duplications (Fig. 2).
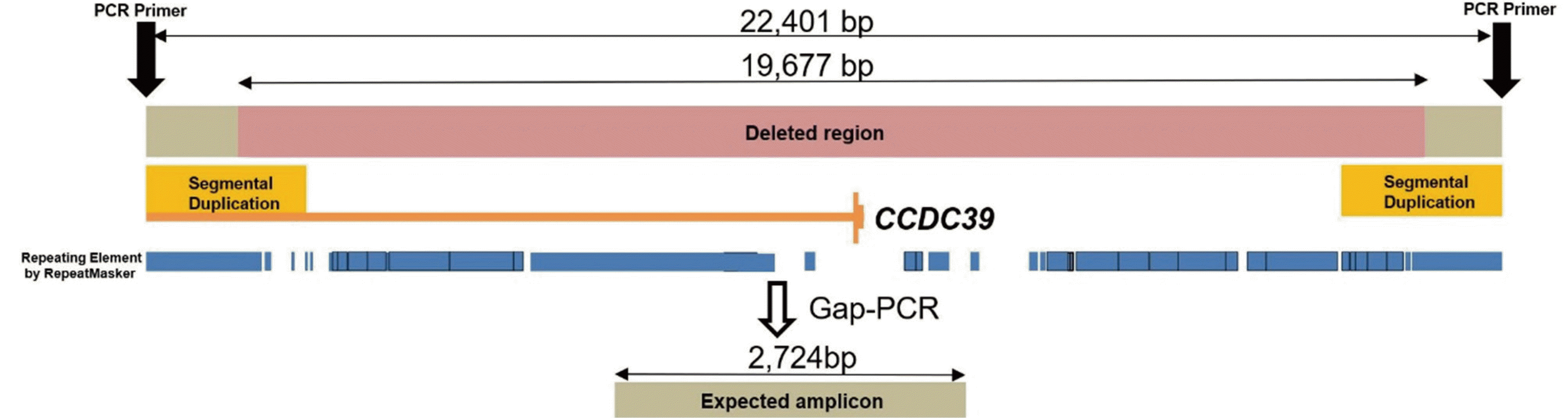 | Fig. 2Schematic representation of the deletion in CCDC39 and gap-PCR. The inferred size of deleted region in CCDC39 is 19,677 bp. PCR primer pairs are designed to flank a deleted region. The expected amplicon size was 2,724 bp. Deleted region (pink bar) includes 5` UTR and exon 1 of CCDC39 (yellow vertical bar). Breakpoints of the deleted region are located repeat regions (blue bar), which make it difficult to confirm with Sanger sequencing.
Abbreviation: UTR, untranslated region.
|
Although early diagnosis of PCD is helpful for treatment and prognosis, there currently is no gold standard diagnostic test. Recently, the diagnosis of PCD using next-generation sequencing-based extended gene panel testing has gained popularity [4, 6]. However, targeted gene panels are limited in detecting noncoding and structural variants [7, 8]. Our finding of a structural variant in CCDC39 through WGS suggests that WGS and gene panel testing play an important role in diagnosing PCD.
In total, 73 disease-causing variants of CCDC39 responsible for PCD worldwide have been reported in the Human Gene Mutation Database; majority are frameshift variants (45.2%), and structural variants, such as exon deletion, are not reported. Loss-of-function of CCDC39 is known to play an important role in disease mechanisms [9]. Deletions that include the 5´ UTR and coding sequences are typically deleterious in loss-of-function genes [10].
In conclusion, we identified a novel structural variant of CCDC39 that causes PCD. We suggest that WGS can play an important role in the diagnosis of PCD owing to structural variants. This is the first report of the CCDC39 variant p.Gln833Valfs*6 in Korea.
Go to : 
 XML Download
XML Download