

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Streptococcus pyogenes causes a wide spectrum of diseases in humans, from mild tonsillopharyngitis, impetigo, and scarlet fever to severe and invasive sepsis, arthritis, necrotizing fasciitis, and streptococcal toxic shock syndrome (STSS) [1]. More than 500,000 deaths due to streptococcal infections are reported worldwide each year, making S. pyogenes a major pathogen associated with high morbidity and mortality [2]. S. pyogenes strains are genetically diverse and have various virulence factors, including adhesion molecules, superantigens, DNases, proteases, and M protein, that are involved in its complex pathogenicity.
S. pyogenes strains can be classified by emm typing that is based on PCR amplification and amplicon sequencing of the emm gene, encoding M protein. Multilocus sequence typing (MLST) based on the amplification and sequencing of seven housekeeping genes and pulsed-field gel electrophoresis (PFGE) of the large genomic fragment have also been used in epidemiological studies [3, 4]. Whole-genome sequencing (WGS) datasets help in discriminating closely related strains and allow epidemiological analysis of small infection clusters. WGS analysis is an ideal molecular typing method for bacteria, as it provides complete genetic information of a strain. Until recently, because of the high cost and technical complexity, WGS was beyond the reach of average diagnostic laboratories [5]. Little has been published on the invasiveness of S. pyogenes strains collected in Korea. Characterization of S. pyogenes in a longitudinal surveillance study of WGS datasets would provide important information about the genomic characteristics, virulence-associated gene profiles, and genomic dynamics during the long period of time.
We genomically characterized S. pyogenes strains collected in Korea between 1997 and 2017 by determining their emm types, MLST-based sequence types (STs), and superantigen gene profiles. We then investigated whether the genomic characteristics of S. pyogenes strains differed based on invasiveness and isolation year.
MATERIALS AND METHODS
Bacterial strain selection
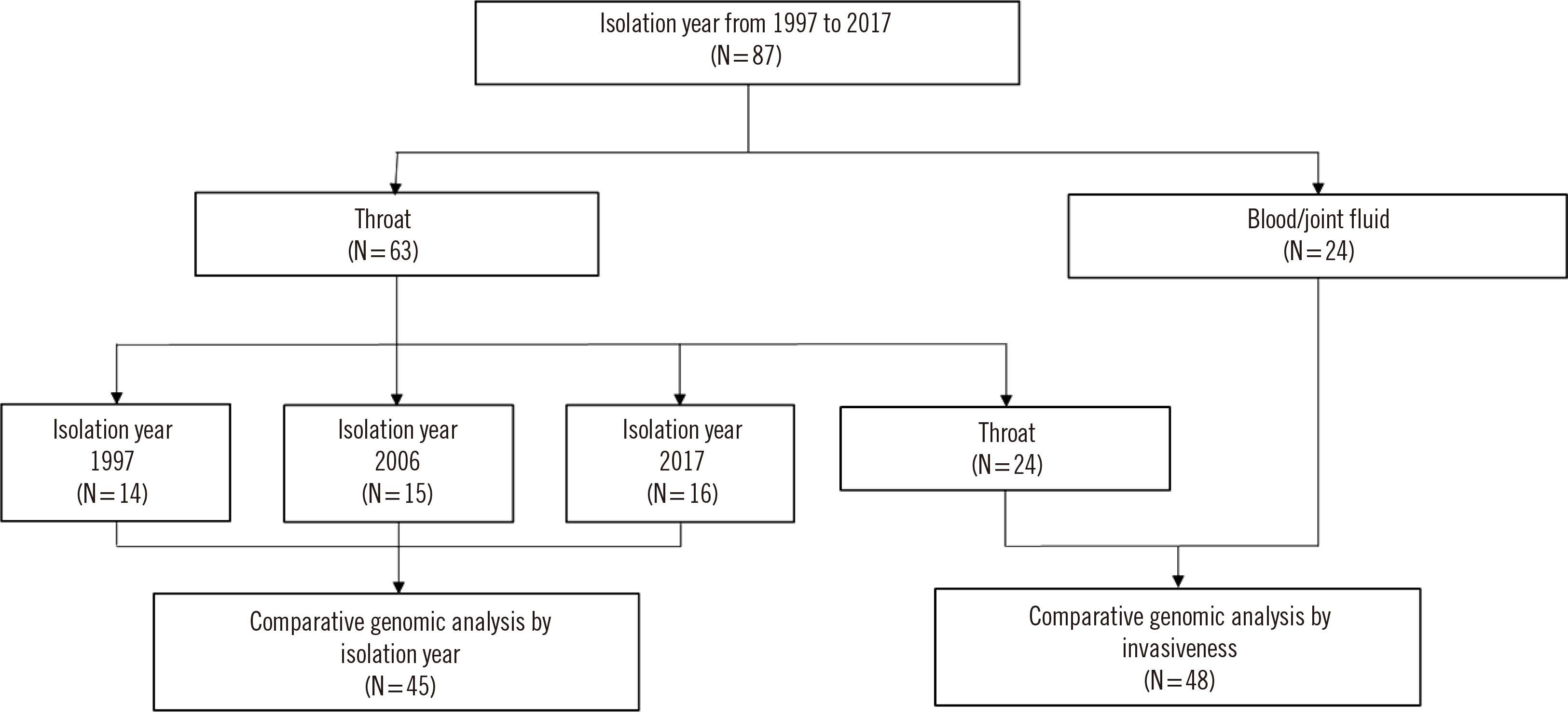
All strains used in this study were collected between 1997 and 2017 and stored in the repository at Gyeongsang National University Hospital (GNUH) in Gyeongnam Province, Korea. Forty-five S. pyogenes strains were selected according to the common emm types in three years: 1997, 2006, and 2017. Forty-eight strains were randomly selected based on their invasiveness between 1997 and 2017. An “invasive strain” was defined as one isolated from a normally sterile body fluid, such as blood, cerebrospinal fluid, pleural fluid, pericardial fluid, joint fluid, bone aspirate, or a deep-tissue abscess [6]. Fig. 1 shows a flow chart of the strain selection procedure. In total, 87 strains were included in this study. For the first analysis to evaluate the genetic evolution over a 20-year time span, non-invasive strains (N=45) were selected every 10 years: 1997, 2006, and 2017. In the second analysis to evaluate assocations between invasiveness and genetic profiles, non-invasive and invasive strains were compared. Sixty-three non-invasive strains were isolated from the throats of carriers who did not have any symptoms or signs of tonsillitis. The other 24 invasive strains were isolated from blood (N=21) or joint fluid (N=3) of patients (Fig. 1).
Bacteria were identified using a Vitek-2 automated identification system (BioMérieux Inc., Marcy l’Étoile, France). All strains were inoculated in 30% glycerol in Todd-Hewitt broth and stored at –70°C. They were recovered on blood agar plates for genetic analysis.
The study protocol was approved by the Institutional Review Board of GNUH (approval number: GNUCH 2018-01-008). Informed consent was waived because of the retrospective nature of the study.
Genomic DNA extraction and WGS
Genomic DNA was extracted using a Wizard Genomic DNA Isolation Kit (Promega, Madison, WI, USA). The DNA and potential culture contamination were checked by 16S rRNA gene sequencing using an ABI 3730 DNA sequencer (Applied Biosystems, Foster City, CA, USA). A draft genome sequence of each strain was generated by MiSeq sequencing (300-bp, paired-end) using a MiSeq Reagent Kit v3 (Illumina, San Diego, CA, USA). Sequencing libraries were prepared using the TruSeq DNA LT sample Prep Kit (Illumina). The Illumina sequencing data were assembled with SPAdes v3.13.0 (Algorithmic Biology Lab, St. Petersburg Academic University of the Russian Academy of Sciences, St. Petersburg, Russia). The EzBioCloud genome database was used for gene finding and functional annotation of the whole-genome assemblies (https://www.ezbiocloud.net). Protein-coding DNA sequences (CDSs) were predicted using Prodigal 2.6.2 [7]. The CDSs were classified based on their roles, with reference to orthologous groups (EggNOG v4.5; http://eggnogdb.embl.de). For more detailed functional annotation, the predicted CDSs were compared with those from the Swiss-Prot (https://www.uniprot.org), KEGG (http://www.genome.jp/kegg/), and SEED (http://pubseed.theseed.org) databases using the UBLAST (https://www.drive5.com/) program.
Comparative genome (CG) analyses
CG analyses comprised two steps (Fig. 1). The first analysis was conducted according to the isolation year (strains of 1997 vs. those of 2006 vs. those of 2017). The second analysis was conducted according to invasiveness (strains from the throat vs. those from blood/joint fluid). CG analysis was conducted by comparing functional genes based on the clustering of orthologous genes. The genome sequences of all strains were obtained from the EzBioCloud database (http://www.ezbiocloud.net/), and average nucleotide identity (ANI) values were calculated. For ANI calculation, the query genomes were cut into small fragments (1,020 bp), and high-scoring pairs between two genome sequences were selected using the USEARCH program (http://www.drive5.com/usearch). Using the calculated ANI values, a dendrogram was constructed using the unweighted pair group method. Homologous regions in a target genome to query open reading frames were determined using the USEARCH program and were aligned using pair-wise global alignment. The matched regions in the subject contig were extracted and saved as homologs [8].
emm genotyping
We used DDBJ Fast Annotation and Submission Tool v1.2.4 (DFAST; https://dfast.nig.ac.jp) for annotation and searched the sequences around the mga annotation encoding multiple virulence gene regulators based on the annotation data [9]. If the sequences around mga were not found, the sequences around the emm1 primer (forward: 5΄-TATT(C/G)GCTTAGAAAATTAA-3΄) were searched throughout the contig sequences using the FASTA format. We extracted the emm sequences between emm1 and emm2 primers (reverse: 5΄-GCAAGTTCTTCAGCTTGTTT-3΄) using the corresponding sequences recovered from the contig data. By inserting the extracted sequences into the Centers for Disease Control and Prevention (CDC) database (https://www2.cdc.gov/vaccines/biotech/strepblast.asp), emm genotypes (including subtypes) were assigned to the extracted sequences.
When sequences were incompletely matched with an emm genotype in the CDC database, we directly PCR-amplified emm using bacterial DNA templates and the emm1/emm2 primer set and sequenced the amplicons after purification using an AccuPrep Purification Kit (Bioneer Corp., Daejeon, Korea). The emm genotypes were assigned directly to the amplified sequences based on the CDC database [10, 11].
Phylogenetic tree and superantigen gene profiling
Phylogenetic analysis was accomplished using ~1.4 million bps of orthologous protein-coding regions for 45 strains according to the isolation year and 48 strains according to invasiveness, respectively (data not shown).
To determine five target genes (speA, speB, speC, ssa, and smeZ) encoding the superantigens (also known as exotoxins), we conducted PCR simulation analysis using the Serial Cloner (http://serialbasics.free.fr/Serial_Cloner.html) application with the contig sequences, as previously reported [12-14]. The primer sets used to amplify the speA, speB, speC, ssa, and smeZ are listed in Table 1. The speB product was included as an internal control in the PCR simulation analysis because all S. pyogenes strains possess the speB sequence (955 bp). Superantigen gene profiles were determined for each strain.
Table 1
Primer sets used to amplify speA, speB, speC, ssa, and smeZ
![]()
MLST
We determined the STs using allelic profiles consisting of seven housekeeping genes (gki, gtr, murI, mutS, recP, xpt, and yqiL) by inserting the contig sequences obtained into the online application MLST v2.0 (https://cge.cbs.dtu.dk/services/MLST/), which is managed by the Center for Genomic Epidemiology at the Technical University of Denmark [15]. The STs were grouped into clonal complexes (CC), whereby related STs were classified as single locus variants, differing in only one housekeeping gene. An expansion of the goeBURST program implemented in PHYLOViZ was used to produce a minimum-spanning tree representing possible relationships among the STs [16].
For novel allelic numbers/STs, we submitted the data (i.e., bacterial genotypic/phenotypic data and patient backgrounds) to the S. pyogenes PubMLST (http://pubmlst.org/organisms/streptococcus-pyogenes) database. The PubMLST curator assigned novel allelic numbers/STs to our strains.
Statistical analysis
We used Fisher’s exact test (two-sided) to determine significant differences in categorical variables, and the chi-square test to compare the proportions in each emm genotype/cluster between invasive and non-invasive strains. SPSS Statistics v22.0 (IBM Corp., Armonk, NY, USA) was used for the analysis. P<0.05 was considered significant.
RESULTS
emm genotypes/clusters
The emm genotypes and cluster types are presented in Supplemental Data Table 1. In total, 21 emm genotypes were identified, with emm1 (emm1.00, emm1.18, emm1.30, and emm1.76), emm4 (emm4.00), and emm12 (emm12.00, emm12.19, and emm12.49) accounting for 19.5%, 13.8%, and 20.7%, respectively. In total, eight emm cluster types were identified, among which the A-C3, A-C4, and E1 types were the most common. Fig. 2 shows the differences among the emm clusters according to invasiveness. There were significantly more invasive strains in cluster A-C3 than in the others (P<0.05). The A-C3 type included the genotypes emm1.0, emm1.18, emm1.3, and emm1.76 (Supplemental Data Table 1, Fig. 2).

ST with goeBURST diagram
The STs are presented in Supplemental Data Table 1. The 87 strains comprised 21 STs with exact loci matched against the PubMLST database. ST36, ST28, and ST39, accounting for 20.7%, 18.4%, and 11.5%, respectively, were the most frequent. There were strong associations of genetic characteristics within the MLST complex. The goeBURST diagram is shown in Fig. 3. There were 17 singletons in the CG analysis, and ST28 showed a clonal distribution of invasive strains in the second analysis. The predominant emm1 lineage belonged to ST28 (Fig. 3).
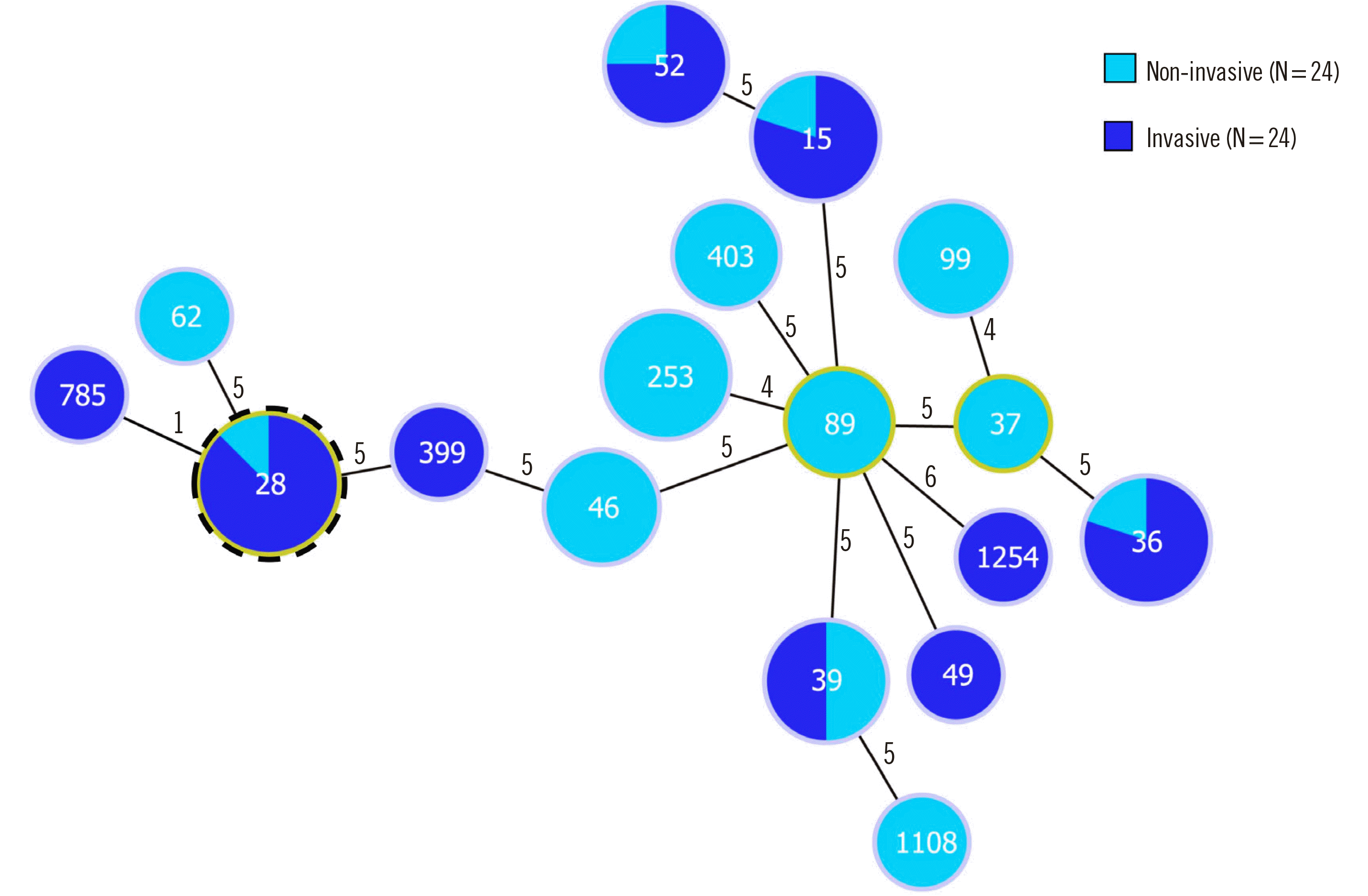
Fig. 3
goeBURST diagram of the relationships among STs according to invasiveness. The numbers in the circles indicate the STs, and the numbers near the lines indicate the number of different alleles between two connected STs. A putative CC is indicated by an outer dotted frame and corresponds to the STs with the highest number of single locus variants. ST785, a single locus variant of ST28, formed CC28.
Abbreviations: ST, sequence type; CC, clonal complex.
![]()
Phylogenetic tree and superantigen gene profiling
The phylogenetic tree based on the periodic comparison showed a sporadic distribution (data not shown). The second analysis revealed the genetic relationships among emm genotypes or STs. Superantigen profiling revealed that speB was present in all strains. speZ-speB and speZ-speB-speC profiles were present in 37.9% and 28.7% of the total strains, respectively. We found no significant association between the coexistence of different superantigen genes and invasiveness.
Virulence-associated CDSs
When comparing gene origins by pan-genome orthologous group (POG) analysis, we found csn1, ispE, nisK, and citC were more significantly present in invasive strains than in non-invasive strains (all P<0.05) (Table 2).
Table 2
Presence or absence of pan-genome orthologous genes according to invasiveness
![]()
We looked for common virulence-associated CDSs among all 87 strains by searching for annotated CDSs based on functional annotation of the whole-genome assemblies. We found 25 CDSs associated with bacterial virulence. Among them, 12 (lactocepin, oleate hydratase, putative glycoslytransferases, capsule biosynthesis protein [CapA], regulatory protein MsrR, internalin-I, deoxyribonuclease, biofilm-regulatory protein, listeriolysin-regulatory protein, streptokinase, C5a peptidase, and M protein) were identified in all strains. CDSs encoding exotoxin A and procollagen-proline 3-dioxygenase were frequently detected in invasive strains (all P<0.05) (Table 3).
Table 3
Comparison of CDSs among all 87 strains by searching annotated CDSs based on functional annotation pipeline of whole-genome assemblies
![]()
DISCUSSION
WGS analyses have proven useful in unraveling the genetic diversity of strains and discriminating between closely related strains. Our study provided information about the genomic characteristics and virulence genes of 87 strains collected in Korea over a 20-year period based on longitudinal analysis of WGS datasets.
Up to 200 emm types have been identified, suggesting that the M protein is a polymorphic protein (https://www.cdc.gov/streplab/index.html). A global review of emm types revealed a total of 205 emm types, including a category of non-typeable strains. The most common emm type was emm1, which accounted for 18.3% of all strains, followed by emm12 (11.1%), emm28 (8.5%), emm3 (6.9%), and emm4 (6.9%) [17]. In Europe, severe clinical manifestations, such as STSS and necrotizing fasciitis, were caused by 45 different types, of which emm1 was the most prevalent, accounting for 37% and 31% of cases, respectively [18]. In Korea, emm1 was significantly more common among invasive cases, whereas emm4, emm6, and emm12 were dominant in non-invasive cases [19].
Globally, emm types influence routine epidemiological surveillance, and MLST is excellent for exotoxin gene profiling [20]. emm1 and emm3 associated with ST28 have traditionally been associated with invasive S. pyogenes strains [19]. In this study, the predominant emm1 lineage belonging to ST28 showed a clonal distribution of invasive strains according to the goeBURST results. ST785, a single locus variant of ST28, also belonged to emm1. The advantages of the conservative approach used by goeBURST, in which links are shown only between STs that differ at a single locus, have been demonstrated by the analysis of meningococcal CCs using goeBURST, which allowed describing the clonal structures of populations in a quantitative way [21].
By searching for virulence-associated CDSs, we found that four genes, csn1 (cas9), ispE, nisK (spaK), and citC, were frequently present in invasive strains. Cas9 is associated with the clustered regularly interspaced short palindromic repeats (CRISPR) array [22]. The type II-A system of S. pyogenes contains four cas genes (cas9, cas1, cas2, and csn1) and six CRISPR spacers targeting a phage endopeptidase, superantigen (sepM), methyltransferase, hyaluronidase, hypothetical protein, and an unknown target. cas9 (previously called csn1) and trans-activating CRISPR RNA are essential for all stages of immunity in the type II-A system [23]. In our study, cas9 was more common in invasive strains. This result indicates the role of cas9 in S. pyogenes pathogenesis and the ability of S. pyogenes to counter external stimuli, while playing a direct role in bacterial immunity.
IspE is involved in the isoprenoid (IPP) biosynthesis pathway. IPPs comprise a large, diverse class of naturally occurring organic chemicals essential for cell survival [24]. The IPP pathway is essential for various vital biological functions of bacteria. IPPs are synthesized via the classical mevalonate pathway or the alternative 2C-methyl-D-erythritol 4-phosphate (MEP) pathway. The distribution of the MEP and mevalonate pathways is highly complex, but there is a clear bias towards the former in pathogenic organisms. In our study, ispE expression was significantly upregulated in invasive strains, suggesting that the IPP biosynthesis pathway is associated with virulence.
Lactococcal nisA is a promoter in the nis cluster that is required for the biosynthesis, immunity, and regulatory systems of S. pyogenes; nisK and spaK also belong to this cluster. The nisA promoter is dependent on NisR and NisK, which are important in the survival mechanisms of S. pyogenes. The nisA promoter allows gene expression modulation in pathogenic streptococci [26]. Bacterial citrate lyase, the key enzyme in citrate fermentation, is encoded by citC. Lactic acid bacteria of the genus Leuconostoc can produce carbon dioxide and C4 aromatic compounds through lactose heterofermentation and citrate utilization [27]. We confirmed that nisR and citC are significantly associated with invasive S. pyogenes. Their protein products are widely found in Lactococcus; therefore, we presume that the genes must have been transmitted via plasmid transfer, allowing efficient control of gene expression by regulatory proteins [28, 29]. The transmitted genes allow S. pyogenes to survive in various environments, strengthening its invasiveness [22].
We investigated virulence-associated CDSs among all 87 strains searched from annotated CDSs based on functional annotation of the whole-genome assemblies. The genes encoding exotoxin A and procollagen-proline 3-dioxygenase were frequently present in invasive strains. Streptococcal exotoxin A is encoded by speA, which is part of bacteriophage T12 [30]. The presence of speA is frequently associated with scarlet fever or rheumatic fever and streptococcal disease [1, 31]. Procollagen-proline 3-dioxygenase catalyzes procollagen L-proline to produce procollagen trans-3-hydroxy-L-proline. This enzyme belongs to the family of oxidoreductases, and its activity has been detected in several strains [32]. A relationship between this enzyme and invasiveness has been rarely observed in S. pyogenes [33].
The phylogenetic analysis revealed no significant associations between the superantigen profiles and invasiveness. Moreover, establishing links between longitudinal groups within the phylogenetic tree was difficult. These results indicate the preservation of stable genetic elements over time. The S. pyogenes population may have maintained a state of host adaptation by maintaining stable genetic elements over long periods [34].
This study had some limitations. Although our study spanned two decades and was population-based, only 87 strains were included, explaining why we did not observe significant genome changes during the study period. We investigated virulence-associated CDSs among all 87 strains by searching only annotated CDSs based on a functional annotation pipeline of whole-genome assemblies rather than by searching the sequences around specific genes or by PCR simulation. We searched for related articles by entering the search terms “Streptococcus pyogenes,” “whole genome,” or “Korea” into the PubMed database (https://pubmed.ncbi.nlm.nih.gov/). However, there were no hits for related manuscripts as of May 26, 2021. This is probably the first report on WGS datasets of S. pyogenes strains from Korea.
In conclusion, this study provided CG characteristics of S. pyogenes according to invasiveness over a 20-year period. Genomic dynamics were stable during this time span. Four genes, csn1, ispE, nisK, and citC, are candidate virulence-associated CDSs in host–pathogen interactions of invasive S. pyogenes strains. Our results showed considerable agreement with previous epidemiological study results, especially regarding the predominant invasive genotypes, i.e., emm1.0, emm1.18, emm1.3, and emm1.76. ST28 showed a clonal distribution of invasive strains. Further epidemiological studies using WGS datasets are needed to better understand and monitor streptococcal virulence.
 XML Download
XML Download