

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Clinicians are confronted with challenges of managing adverse effects of conventional opioid analgesics such as itching, constipation, nausea/vomiting, and respiratory/cardiovascular depression. Although some tolerance to opioid-induced adverse effects develops over time, concerns of euphoria, abuse, and addiction persist. Hence a keen interest in the model of “ligand bias” at G protein receptors has been perceived to preferentially stimulate single intracellular signaling pathway to produce the safer, more-effective, and better-tolerated drug [1].
Guanine nucleotide binding protein-coupled receptors (GPCRs) are 7-transmembrane (7 TM, heptahelical) receptors that bind to agonists and couple to G proteins resulting in activation of complex intracellular signaling pathways [1]. Various µ-opioid receptor (MOR) agonists can activate one or a number of downstream pathways of the GPCR at different intervals: G protein-dependent or independent signaling, desensitization, and endocytosis [2]. Bias or selectivity to the downstream pathways involving the GPCR may provide improved analgesia and decreased adverse reactions (ADRs). The five main super-families of GPCRs classification consist of glutamate, rhodopsin, adhesion, frizzled/taste 2, and secretin, forming the GRAFS classification system [345].
Peripherally-acting opioids, such as D-Ala(2), N-MePhe(4), Gly(5)-ol-enkephalin (DAMGO), acting neither centrally (supra-spinal) nor spinally, drew significant attention by achieving adequate analgesia while reducing ADRs and tolerance development [67].
A formulation containing opioid agonist/antagonist in a fixed 2:1 ratio has been introduced to reduce ADRs, especially opioid-induced constipation [8]. However, at the equivalent dose, the combined formulation often failed to provide equal analgesia as the conventional opioid [9]. It is not uncommon for patients to complain of inadequate pain relief and request the traditional single opioid agonist. Hence, there is a clear need for a new novel analgesic agent devoid of undesirable side effects while maintaining analgesic efficacy.
This review article discusses a brief history, pharmacokinetics and pharmacodynamics, ADRs of the new opioid oliceridine (TRV130, Olinvo®, Trevena, Inc, King of Prussia, PA) with a focus on the unique mechanism of action on the µ receptor G protein pathway selective (µ-GPS) modulator.
Go to : 
MAIN BODY
1. Brief history
GPCRs have become a focus in drug development based on the concept of ligand bias or functional selectivity [10]. This concept originated from an animal study of ADRs to morphine in β-arrestin 2 knockout mice. The mice lacking the G protein-coupled receptor regulatory protein, β-arrestin 2 (β-arrestin 2 knockout mice), showed enhanced and prolonged morphine analgesia while mitigating morphine tolerance, respiratory depression, and constipation in three previous studies from 1999 to 2005 [111213].
The first human study of oliceridine in healthy volunteers (phase I study), evaluated drug tolerability, pharmacokinetics, and pharmacodynamics in 2014 [14]. In this study, oliceridine was well-tolerated over a dose range from 0.15 to 7 mg administered intravenously over 1 hour. The geometric mean exposure and Cmax were dose-linear with a half-life of 1.6-2.7 hours. The drug clearance was reduced by 53% in cytochrome P450 2D6 (CYP2D6)-poor metabolizers. The results showed a dose- and exposure-related pupil constriction (miosis), confirming central compartment MOR engagement. Nausea and vomiting were observed at a dose of 7 mg.
A comparative trial of oliceridine, using healthy volunteers, was performed with placebo and morphine by drug developer (Trevena Inc) in 2014 to measure safety, tolerability, and analgesia. This study showed that 3 mg and 4.5 mg of oliceridine elicited higher peak analgesia with faster onset action than 10 mg of morphine with similar duration. In addition, ADRs such as respiratory drive reduction and nausea was less than with morphine [15].
Phase II clinical trial of oliceridine in 2016 evaluated a pharmacokinetic (PK), pharmacodynamic (PD), and dose simulation to develop a mathematical model based on post-operative pain relief. Across two phase 2 studies of patients with pain following hard- and soft-tissue surgeries, oliceridine led to greater early reductions in pain intensity than morphine. Following the abdominoplasty, oliceridine was associated with a lower percentage of patients with nausea (41% and 46% with 0.1 mg and 0.35 mg, respectively) than the morphine group (72%; P < 0.05 for both comparisons). Oliceridine also was associated with a lower percentage of patients with vomiting (15% for both 0.1 mg and 0.35 mg) than the morphine group (42%; P < 0.05 for both comparisons). No drug-related serious adverse events were reported. These results suggest that oliceridine may have an improved gastrointestinal tolerability profile compared to morphine [1617181920].
The two phase III APOLLO pivotal efficacy studies, intravenous oliceridine showed statistically superior analgesia than the placebo in moderate-to-severe acute pain following bunionectomy and abdominoplasty, while demonstrating improved respiratory safety and gastrointestinal tolerability compared to morphine. Hence oliceridine may be highly effective and well-tolerated for patients in need of strong analgesia postoperatively [21].
Subsequently, Trevena, Inc. has submitted New Drug Application for oliceridine injection to the U.S. Food and Drug Administration in November 2017. The submission includes data showing that intravenous oliceridine demonstrated analgesic efficacy in all three dosing regimens (0.1 mg, 0.35 mg, 0.5 mg). The filing also includes safety and tolerability data from phase 2 and phase 3 studies, including the ATHENA open-label safety study. Additional pharmacokinetic data, clinical pharmacology data, and results from five randomized controlled trials with head to head comparisons to morphine support potential differentiation of oliceridine. The company expects oliceridine to be a Schedule II controlled substance [21].
Currently known biased MOR agonists for GPCRs are oliceridine, herkinorin, PZM21, and mitragynine, which exhibit decreased ADRs from opioids [22].
2. Mechanism of action
In human, the largest family of over 800 genes encodes ubiquitous receptor proteins with a unique seven-transmembrane (7 TM) configuration [23]. The 7 TM receptors are commonly referred to as GPCRs which utilize heterotrimeric protein signaling and are the most common target of the therapeutic drugs.
The 7 TM receptors have been classified into three families: A, B, and C. The largest family, A, includes rhodopsin (for light), adrenergic receptors (for blood pressure), olfactory receptors (for smell), as well as opioid receptors. Family B includes the gastrointestinal peptide hormone family secretin, glucagon, vasoactive intestinal peptide (VIP), growth hormone, corticotropin-releasing hormone, calcitonin, and parathyroid hormone. Family C consists of the metabotropic glutamate receptor family the gamma amino butyric acid B (GABAB), the calcium-sensing, and taste receptors [23].
Many of the commonly used therapeutic drugs in clinical practice act via GPCR. Some examples are: 1) fexofenadine, histamine 2 (H1) antagonist, for allergies, 2) valsartan, angiotensin 1 (AT1) antagonist, for hypertension, 3) famotidine, histamine 2 (H2) antagonist, for gastric ulcer, 4) sumatriptan, 5-hydroxytryptamine 1D (HT1D) agonist, for migraine, 5) leuprorelin, luteinizing hormone-releasing hormone (LH-RH) agonist, for cancer, 6) gabapentin, GABAB agonist, for neuropathic pain, 7) clopidogrel, P2Y12 [the Gi coupled platelet receptor for adenosine diphosphate (ADP)] antagonist, for stroke, 8) risperidone, mixed 5-HT2/dopamine 2 (D2) antagonist, for schizophrenia, 9) salmeterol, β1 agonist, for asthma, and 10) olanzapine, mixed 5-HT2/D1/D2 antagonist for schizophrenia [24].
There are at least three essential structures which govern G protein signaling: 1) GPCR, 2) heterotrimeric G protein, and 3) the target protein (effector). The G protein, guanine nucleotide binding protein has three different subunits (Gα, β, and γ), a heterotrimeric structure, in an active state. In the resting state, the α subunit is bound to guanosine diphosphate (GDP) and is associated with the Gβγ subunits. Upon binding a ligand, the GPCR is activated. The active GPCR increases guanosine triphosphate-guanosine diphosphate (GTP-GDP) exchange on the G protein, and the active GTP-bound form of Gα dissociates from the Gβγ subunits. These two separated GTP-Gα and Gβγ subunits activate their respective effectors. However, upon withdrawal of the ligand from the GPCR, when the GPCR is no longer active, the intrinsic GTPase activity of the Gα subunit hydrolyzes the GTP to GDP. The GDP-bound, inactive, Gα binds to the Gβγ again [2526].
It is essential to understand the mechanism of oliceridine to discern the differences between the opioid agonist, antagonist, G protein biased ligand, and β-arrestin biased ligand. A classic opioid agonist nonselectively activates both G protein and β-arrestin-mediated signaling. On the contrary, a traditional opioid antagonist activates neither G protein nor β-arrestin-mediated signaling. Interestingly, a G protein biased ligand promotes G protein signaling without β-arrestin mediated desensitization, internalization, or signaling. On the other hand, a β-arrestin biased ligand promotes β-arrestin mediated desensitization, internalization, and signaling in the absence of G protein activation [27].
There is activation of at least 4 different downstream pathways from the MOR agonist binding to the receptor: 1) G protein dependent signaling: regulation of ion (calcium and potassium) channels and inhibition of adenylyl cyclase, 2) G protein independent signaling: β-arrestin (β-arr), extracellular signal regulated kinase (ERK or Erk), and c-Jun N-terminal kinase (JNKs), 3) desensitization: G protein-coupled receptor kinase (GRK), β-arr, protein kinase C (PKC) and protein kinase A (PKA), and 4) endocytosis: clathrin, γ-arr, dynamin (Dyn), phospholipase 2 (PLD2), and assembly polypeptide 2 (AP2) [2].
The current understanding of GPCR pathways, focused on phosphorylation/ dephosphorylation, includes 1) agonist binding and G protein activation [increased K+ outward and Ca2+ inward currents with decreased cyclic adenosine monophosphate (cAMP) by inhibition of adenylyl cyclase], 2) receptor phosphorylation, 3) arrestin binding, 4) clustering in clathrin-coated pits (CCPs) and endocytosis (clathrin-dependent endocytosis), 5) receptor dephosphorylation, and 6) recycling. The G protein biased ligand promotes G protein activation but inhibits β-arrestin binding. Efficient resensitization after desensitization of MORs reduces the development of tolerance. Resensitization is also very efficient in the absence of endocytosis. Rapid and strong internalization of opioid agonists produces less tolerance (Fig. 1) [2829].
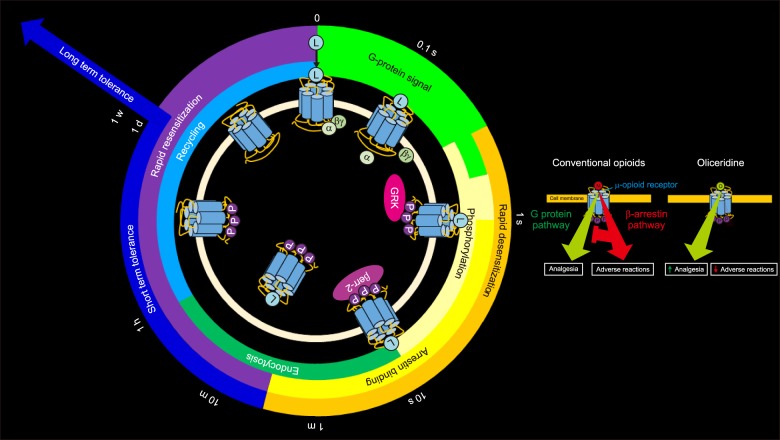 | Fig. 1The µ-opioid receptor regulation (recycling) with a µ receptor G protein pathway selective (µ-GPS) modulator, oliceridine (TRV 130) and a µ-opioid receptor agonist, morphine. 1) Immediately after µ-opioid receptor agonists and µ-GPS modulators bind to the µ-opioid receptors, the G protein is activated (increased K+ outward and Ca2+ inward currents with decreased cAMP by inhibition of adenylyl cyclase), 2) receptor phosphorylation, 3) arrestin binding, 4) clustering in clathrin-coated pits (CCPs) and endocytosis (clathrin-dependent endocytosis), 5) receptor dephosphorylation, and 6) recycling. The G protein biased ligand (µ-GPS modulators) promotes G protein activation, but inhibits β-arrestin binding. On the contrary, the classic opioid receptor agonist, morphine, increases both G protein activation and β-arrestin binding. The β-arrestin binding not only increases adverse reactions but also decrease G protein activation-related analgesic effect. This schematic diagram also shows desensitization, followed by short term and long term tolerance. L: ligand, M: opioid, M: morphine (Modified from Kliewer A, Reinscheid RK, Schulz S. Emerging paradigms of G protein-coupled receptor dephosphorylation. Trends Pharmacol Sci 2017; 38: 621-36 [28]. Dang VC, Christie MJ. Mechanisms of rapid opioid receptor desensitization, resensitization and tolerance in brain neurons. Br J Pharmacol 2012; 165: 1704-16 [29].).
|
Desensitization is defined as a progressive reduction of signal transduction that occurs after opioid receptor activation depending on the agonist and the signaling pathway. The rapid desensitization is regulated by the ion channel conductance, while the sustained desensitization is regulated by enzymes such as adenylyl cyclase and mitogen-activated protein (MAP) kinases. Mechanisms of desensitization appear to share common pathways: phosphorylation, the involvement of arrestin, endocytosis, and receptor trafficking. Moreover, the desensitization process will dependent on agonist (biased agonism), time exposure, cell system, and receptor. Tolerance is defined as a decrease of the drug response due to the reduction of the outward current of potassium of the MOR [30].
3. Pharmacodynamics and pharmacokinetics
1) Pharmacokinetics
Oliceridine is primarily metabolized by cytochrome P450 2D6 (CYP2D6) and CYP3A4 in human hepatic microsomes. It reaches peak plasma concentrations within 10 m after single bolus injection and at the end of the 1-h continuous infusions.
Its concentration declines in a biphasic manner, indicating rapid distribution followed by an elimination phase. The half-life is from 1.6 to 2.7 h after continuous infusion over 1 h, which is similar to those of morphine and hydromorphone. The mean clearance ranges from 34 L/h and decreases linearly when increasing the dose [1415].
2) Pharmacodynamics
Objective pupil constriction, confirming central compartment MOR engagement, may express a subjective analgesic effect. Oliceridine elicits marked pupil constriction (miosis), which lasts for at least 2 hours after the infusion is discontinued. Peak miosis occurs at 10 minutes after infusion, though this is dose-dependent. Oliceridine, ranging from 1.2 to 4.0 mg, elicits pupil constriction of 0.4 to 2.7 mm, which is similar to doses of intravenous morphine (2 to 8 mg) and buccal fentanyl (100 to 400 µg) [14].
Oliceridine 2 and 3 mg in every 3 hours produced significant pain relief within 5 minutes of acute pain after bunionectomy, similar to morphine 4 mg every 4 hours [21]. The dose-dependent analgesic effect to the hand removal latency test (cold water pain test) for a single intravenous injection showed in 105 and 116 seconds for 3 and 4.5 mg of oliceridine, respectively, compared to at a 75 seconds latency for 10 mg of morphine [15].
Frequent ADRs, at a dose of 7 mg of continuous infusion of oliceridine over 1 hour are nausea, vomiting, pruritus, hyperhidrosis, dizziness, headache, somnolence, feeling hot, and a feeling of relaxation in healthy volunteers [14].
Respiratory depression, the most dreadful ADR, was noted after a single intravenous bolus injection of oliceridine for the first hour at 1.5 mg, and for the first two hours at 3 and 4.5 mg. However, respiratory drive reduction from any dose of oliceridine (1.5, 3, and 4.5 mg) was less than that of 10 mg of morphine [15].
Severe nausea after a single bolus injection was less commonly noted in the group who received 1.5 and 3 mg of oliceridine than in the group who received 10 mg of morphine. However, 4.5 mg of oliceridine showed a more frequent incidence of severe nausea than 10 mg of morphine [15].
In conclusion, oliceridine, compared with morphine, produced greater analgesia at 5 or 10 minutes after a bolus injection or continuous infusion with less respiratory depression and severe nausea.
4. Adverse reactions
The most frequently reported ADRs were nausea, dizziness, headache, vomiting, somnolence, constipation, flushing, hot flush, pruritus, dry mouth, and feeling hot in a dose-dependent fashion from 0.5 mg to 3 mg in every 3 h. The ADRs in the group receiving 2 mg injections of intravenous oliceridine every 3 h were similar to those in the group receiving 4 mg of intravenous morphine every 4 h [21].
Go to :
CONCLUSIONS
The launching of oliceridine, a novel µ receptor G protein pathway selective (µ-GPS) modulator, is a step in the right direction to maximize benefits while minimizing adverse effects of opioid. However, a long-term safety data need to be carefully assessed before the new drug release to avoid the acrimonious mistake of the selective cyclooxygenase-2 (COX-2) inhibitor rofecoxib. The much fanfare of COX-2 selective NSAIDs was fewer gastrointestinal adverse effects, but the incidence of heart attack and stroke were increased compared to the nonselective NSAIDs [31].
During the process of opioid-mediated intracellular signaling, classic opioid agonists bind and activate opioid receptors. The activation of opioid receptors results in a dissociation of G-protein heterotrimers. The dissociated Gα1 subunit inhibits adenylyl cyclase results in cAMP inhibition. The dissociated Gβγ subunit increases the K+ outward flow and decreases the Ca2+ inward flow. This process causes inhibition of neuronal excitability and neurotransmitters resulting in analgesia. On the contrary, β-arrestin binds to the phosphorylated MOR and results in opioid receptor internalization, desensitization, tolerance, and ADRs [32].
A potent, selective and G protein biased MOR agonist, oliceridine, shows enhanced efficacy and duration of pain relief with reduced ADRs, leading to a better therapeutic index. Another selective Gi-based µ opioid agonist with minimal β-arrestin-2 recruitment, PZM21, has recently been discovered and has potent κ-opioid receptor (KOR) antagonist activity [3334].
There will be a general trend of potentiating G-protein MOR signaling while reducing the undesirable ADRs of opioids via the β-arrestin pathway in future studies. Clinicians wait launching of this novel opioid while trusting unpredictable ADRs will not surface.
Go to :
 XML Download
XML Download