

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Patients with muscle pain, headaches, or functional gastrointestinal disorders are approximately 2.5-10 times more likely to be screened with a positive match for a pain disorder, generalized anxiety disorder, or major depressive disorder in a primary care setting [1]. Anxiety is a recognized symptom of many psychiatric disorders, including generalized anxiety disorder (GAD), social anxiety disorder (SAD), obsessive-compulsive disorder (OCD), and posttraumatic stress disorder (PTSD) [2].
Several tracts carry the nociceptive signals to the brain. In addition to the spinothalamic tract, spinoreticular tract, spinohypothalamic tract, and cervicothalamic tract, the spinomesencephalic tract projects to the mesencephalic reticular formation and the periaqueductal gray matter. Neurons from the spinomesencephalic tract synapse with neurons that terminate in the amygdala, which involves emotions and a fear-based response [3,4].
The somatosensory cortex from thalamic nucleus is charged in localization of painful stimuli. However, projections to the limbic system trigger the emotional response to pain. Projections to the insular cortex help modulate the autonomic response and integrate sensory, affective, and cognitive responses to pain [5].
The spinal pathways to limbic structures and medial thalamic nuclei provide direct inputs to brain areas involved in affect. Another source is from the spinal pathways to somatosensory thalamic and cortical areas and then through a corticolimbic pathway. Both direct and corticolimbic pathways converge on the same anterior cingulate cortical and subcortical structures whose function may be to establish emotional valence and response priorities [6,7].
Benzodiazepine anxiolytics have played a central role in the pharmacologic management of anxiety disorders for about 50 years. These agents reduce anxiety rapidly by allosterically enhancing the postsynaptic actions of gamma-aminobutyric acid (GABA) at inhibitory type A GABA receptors, but adverse effects limit their use in chronic anxiety disorders.
Selective serotonin reuptake inhibitors and selective serotonin/norepinephrine reuptake inhibitors have emerged as an effective first-line alternative treatment of such anxiety disorders. However, many individuals are non-responsive and adverse effects can be limiting.
Research into a relatively new class of agents known as neurosteroids has revealed novel modulatory sites and mechanisms of action that are providing insights into the pathophysiology of certain anxiety disorders, potentially bridging the gap between the GABAergic and serotonergic circuits underlying anxiety [2].
Using the PubMed search engine to access the MEDLINE database of references, a total of 36 studies were found related to the keyword, "etifoxine". Eight studies, which were not written in English or not found, were excluded. In the following review, the topics of action mechanisms, pharmacodynamics and pharmacokinetics, and clinical application of etifoxine - a non-benzodiazepine anxiolytic - will be introduced and focused on pain patients with anxiety.
Go to : 
ACTION MECHANISMS FOR ANXIOLYSIS
The exact action mechanism of etifoxine (6-chloro-2-ethylamino-4-methyl-4-phenyl-4H-3, 1-benzoxazine hydrochloride) is not fully understood yet. The known mechanism of etifoxine is a direct potentiation of GABAA receptor activation though a site different from the classical benzodiazepine binding motif [8]. It also modulates GABAA receptors via stimulation of neurosteroid production after the binding of etifoxine to the 18 kDa translocator protein (TSPO) of the outer mitochondrial membrane, previously known as the peripheral benzodiazepine receptor (PBR) [9,10,11] (Fig. 1).
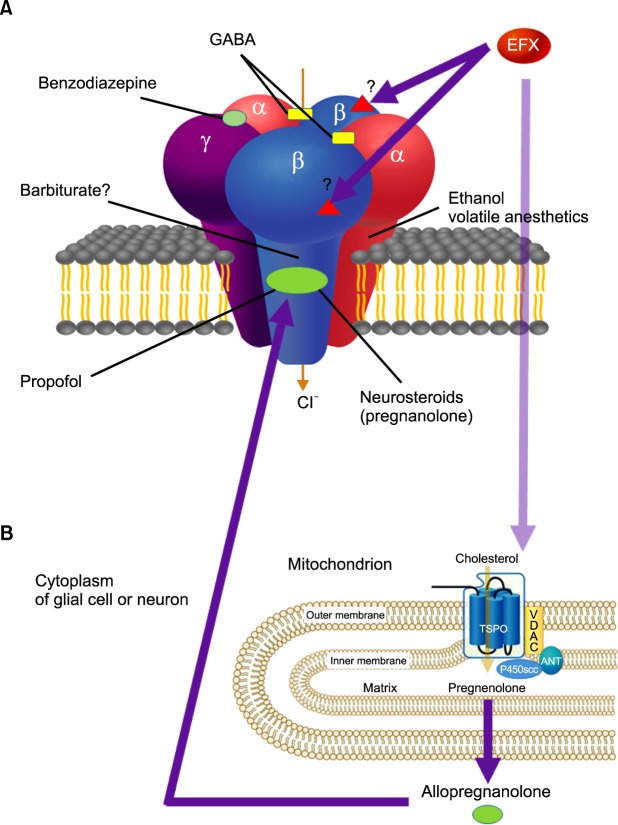 | Fig. 1Schematic action mechanism of etifoxine (EFX). Hypothetical schematic model of the GABAA receptor is a pentameric structure, with the five subunits (two α, two β, and a single γ subunit) arranged around a central chloride-selective pore. A variety of chemical compounds are capable of acting on GABAA receptors to modulate its channel function. The receptor has specific sites for each chemical compound (benzodiazepines, barbiturates, and neurosteroids), which are allosteric sites for modulation of GABA currents or chloride conductance. Etifoxine is a direct potentiation of GABAA receptor activation though a site different from the classical benzodiazepine binding motif. (A) Presumably etifoxine appears to produce its anxiolytic effects by binding to β2 and β3 subunits of the GABAA receptor complex. The effects of etifoxine are not reversed by the benzodiazepine antagonist flumazenil. (B) In addition, etifoxine modulates GABAA receptors via stimulation of neurosteroid production. This occurs through the binding of etifoxine to the 18 kDa translocator protein (TSPO) of the outer mitochondrial membrane, known as the peripheral benzodiazepine receptor (PBR). TSPO or PBR interacts with a voltage-dependent anion channel (VDAC, a protein that is present in outer mitochondrial membrane - inner mitochondrial membrane contact sites) and the adenine nucleotide transporter (ANT, in inner mitochondrial membrane) to form a complex. Cholesterol transport across the outer mitochondrial membrane through TSPO is activated by etifoxine binding to the protein. The cholesterol side-chain-cleaving cytochrome P450 enzyme (P450scc), which is located at the inner mitochondrial membrane, converts cholesterol to pregnenolone, which is further metabolized through several steps by enzymes present in the endoplasmic reticulum and finally converted into neurosteroid allopregnanolone (ALLO). ALLO acts in an autocrine and paracrine manner and are potent positive allosteric modulators of synaptic and extrasynaptic GABAA receptor. They also modulate GABAA receptor function through a binding site different from that of benzodiazepines (adapted from Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, et al. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov 2010; 9: 971-88).
|
1. Direct potentiation of GABAA receptor containing the β2 or the β3 subunit activation
GABAA receptors are hetero-pentameric ligand-gated chloride channels and integral membrane proteins. They are the major inhibitory neurotransmitter receptors in mammalian brain. Each isoform consists of 5 homologous or identical subunits surrounding a central chloride ion-selective channel gated by GABA. The 19 known genes coding for human GABAA receptor subunit are α1-6, β1-3, γ1-3, δ, ε, θ, π, and ρ1-3. The chromosomes related to GABAA receptors are 1, 3, 4, 5, 6, 15, and X. The major isoform GABAA receptors, two α1- two β2- one γ2, are grouped around a central ion pore. The 2 binding sites of GABA are located at the interfaces between α1 and β2.
The GABAA receptors are responsive to a wide variety of drugs, such as benzodiazepines, barbiturates, intravenous and inhalational anesthetics, and non-benzodiazepine anxiolytics. Benzodiazepines are binding at the interfaces between α1 and γ2 subunits [12]. The binding sites for barbiturate, inhalational anesthetics, and ethanol are the same as those of GABA, at the interfaces between α1 and β2 [13,14]. Propofol and neurosteroids are binding to the β2 subunit [15]. However, the binding sites of GABAA receptors with various anxiolytic, sedative, and hypnotic agents are still uncertain. They show dose-dependent therapeutic effects, from anxiolytic, to sedative, and finally to hypnotic effects.
Etifoxine is known to act preferentially on GABAA receptors containing the β2 or the β3 subunit. Its potentiating effects should therefore be maximized in the brain areas where these subunits predominate over the β1 isoform. Further studies will be required to determine what amino acid residues underlie the preference of etifoxine for β2/3-containing receptors [8].
2. Modulation of GABAA receptors via stimulation of neurosteroid production through the binding of etifoxine to the 18 kDa translocator protein
The TSPO is localized primarily in the outer mitochondrial membrane of steroid-synthesizing cells, including those in the central and peripheral nervous system. The TSPO, previously known as the peripheral benzodiazepine receptor (PBR), is a widely distributed transmembrane protein that is located mainly in the outer mitochondrial membrane. The PBR binds to high-affinity drug ligands and cholesterol. The main functions of the PBR include; 1) the regulation of cholesterol transport and the synthesis of steroid hormones, 2) porphyrin transport and heme synthesis, 3) apoptosis, 4) cell proliferation, 5) anion transport, 6) regulation of mitochondrial functions, and 7) immunomodulation.
Based on these functions, there are many potential clinical applications of PBR modulation, such as in oncologic, endocrine, neuropsychiatric, and neurodegenerative diseases. Although PBR is a widely used and accepted name in the scientific community, recent data regarding the structure and molecular function of this protein increasingly support renaming it to more accurately represent its subcellular roles and putative tissue-specific functions. Therefore, TSPO is proposed as a new name, regardless of the subcellular localization of the protein since 2006 [10].
One of its main functions is the transport of the substrate cholesterol into mitochondria, a prerequisite for steroid synthesis, including the highly effective GABAA receptor modulator pregnenolone [16]. The cytoplasmic pregnenolone from the mitochondria converts into progesterone, 5α-DHPROG, and finally 5α3α-THPROG. The 5α3α-THPROG modulates GABAA receptor and increases inward-flow of the chloride into the cytoplasm [17].
In conclusion, etifoxine potentiates GABAA receptor-function not only by a direct allosteric effect but also by activation of TSPO indirectly [9].
Go to :
ACTION MECHANISMS FOR PERIPERAL NERVE HEALING AND REGENERATION
Etifoxine, as a ligand of the TSPO, is proven to promote axonal regeneration, to modulate inflammatory response, and to improve functional recovery since 2008 [18,19]. It also stimulates neurite outgrowth and glia-derived neurite outgrowth [20,21]. Therefore, it provides benefits in nerve repair with acelluar nerve grafts [22]. However, more studies are needed for the application of clinical practice, including multiple sclerosis [23].
Go to :
ACTION MECHANISMS FOR REDUCING NEUROPATHIC PAIN
A study of etifoxine related to reduction and prevention of vincristine-induced neuropathic pain symptoms was released in 2009. Chemotherapy-related pain relief by etifoxine results from 3α-reduced neurosteroids based on TSPO [24]. Etifoxine also stimulates allopregnanolone synthesis in the spinal cord to produce analgesia in experimental mononeuropathy [25]. It can reduce pain in experimental monoarthritis by combined actions that protect spinal inhibition and limits central inflammatory processes. These processes include activation of microglia and spinal disinhibition. It is not only amplified spinal GABAergic inhibition, but also prevented prostaglandin E2-induced glycinergic disinhibition and restored a normal spinal pain processing [26]. However, all 3 studies were performed by the same researchers and needed clinical verification.
Go to :
PHARMACOKINETICS AND PHARMACODYNAMICS
1. Pharmacokinetics
After oral administration, etifoxine is rapidly absorbed by the gastrointestinal tract. Time to reach maximal concentration in the blood is 2-3 hours. It is metabolized rapidly in the liver to form several metabolites. One of the metabolites, diethyl-etifoxine, is active. It can also cross the placental barrier. Its half-life is about 6 hours and half-life of its active metabolite is almost 20 hours. It is mainly excreted in the urine as metabolites, and also excreted in the bile. Small amounts are excreted in unchanged form.
2. Pharmacodynamics
The usual dose of etifoxine is 50-200 mg/d for no longer than 12 weeks. Symptoms of overdose are lethargy and excessive sleepiness. It can potentiate the other drugs which can depress the central nervous system. It also amplifies the effects of alcoholic beverages. The most common adverse effect is drowsiness in the early days of treatment. It is contraindicated in the state of shock, myasthenia gravis, severe hepatorenal disorders, adolescents under 18 years old, hypersensitivity (skin rash, urticaria, or angioedema), and galactosemia.
Go to :
CLINICAL APPLICATION
Before prescribing anxiolytics to patients with anxiety, it is essential to evaluate anxiety rating scales.
1. Anxiety rating scale
The Hamilton anxiety rating scale (HAM-A) was one of the first rating scales developed to measure the severity of anxiety symptoms by clinicians and is still used today in both clinical and research settings. This scale is composed of 14 items (anxious mood, tension, fears, insomnia, intellectual symptoms, depressed mood, somatic [muscular] symptoms, somatic [sensory] symptoms, cardiovascular symptoms, respiratory symptoms, gastrointestinal symptoms, genitourinary symptoms, autonomic symptoms, and behavior at interview) with 5 grades of responses (0 = not present, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe). The interpretation about the summation of each score can be divided as mild (0-17), mild to moderate (18-24), and severe (25-30) anxiety [27].
Other anxiety-related rating scales, accessible free of charge online, are generalized anxiety disorder 7 items (GAD-7), Liebowitz social anxiety scale (LSAS), pain and agoraphobia scale (PAS), Spence children's anxiety scale (SCAS), social phobia inventory (SPIN), Taylor manifest anxiety scale (TMAS), and Zung self-rating anxiety scale (SAS).
2. Clinical applications for pain patients with anxiety
Benzodiazepines are widely used in the treatment of anxiety, insomnia, induction and maintenance of anesthesia as well as for epileptic disorders. However, they have a common adverse effect, such as dose-related anterograde amnesia, sedation, and impaired psychomotor performance. A comparison study revealed that while vigilance, psychomotor performance and free recall were significantly impaired by lorazepam (2 mg), neither dosage of etifoxine (50 and 100 mg) produced such effects. In another study, etifoxine (150 mg/day) showed a better anxiolytic effect and memory recall with less withdrawal symptoms than lorazepam (0.5-1 mg/day) [30,31].
Etifoxine attenuates stress-induced corticotropin-releasing hormone (CRH) activation. Therefore, it may be helpful in the treatment of selected anxious patients with altered autonomic symptomatology [32].
Etifoxine should be avoided when consuming alcoholic beverages. Reports of etifoxine-related acute hepatitis exist [33].
In conclusion, etifoxine are used for various emotional and bodily reactions followed by anxiety. It is contraindicated in situations such as shock, severely impaired liver or kidney function, and severe respiratory failure. The average dosage is 150 mg per day for no more than 12 weeks. The most common adverse effect is drowsiness at the initial stage. It does not usually cause any withdrawal syndromes.
Go to :
CONCLUSIONS
Many painful syndromes including myofascial pain syndrome, fibromyalgia, and sympathetic maintained pain syndromes, are commonly combined with anxiety [34,35]. Etifoxine shows less adverse effects of anterograde amnesia, sedation, impaired psychomotor performance, and withdrawal syndromes than those of benzodiazepines. It potentiates the GABAA receptor-function by a direct allosteric effect and by an indirect mechanism related to the activation of TSPO. Non-benzodiazepine anxiolytics, etifoxine, will replenish shortcomings of benzodiazepines and selective serotonin reuptake inhibitors according to animated studies related to TSPO. In addition, further studies are required for the roles in the peripheral nerve regeneration and relief of neuropathic pain.
Go to :
 XML Download
XML Download