

 PDF
PDF Citation
Citation Print
Print


Malignant peripheral nerve sheath tumors (MPNSTs) are very rare sarcomas derived from various cells in the peripheral nerve sheath. Although MPNSTs can occur accidentally in healthy people, these tumors often occur in patients with neurofibromatosis Type 1 (NF 1) [1]. The prognosis of MPNSTs is worse in patients with NF1 than in patients without NF 1 [2]. It usually occurs near a nerve trunk; therefore, the most common anatomic sites for MPNSTs are the extremities, the trunk, the head and neck [1,3,4]. If a patient does not have specific signs and symptoms except for pain, an early diagnosis of MPNSTs is difficult. Computer tomography (CT) scanning, magnetic resonance image (MRI) and needle biopsy may be useful in the diagnosis of MPNSTs. The treatment for MPNSTs is wide surgical resection with or without adjuvant radiotherapy or chemotherapy. Herein, we report a case of concurrent malignant peripheral nerve sheath tumor in a patient with complex regional pain syndrome (CRPS) type 1.
CASE REPORT
A 44-year-old female patient was referred to our pain clinic due to persistent pain in the lateral side of the left ankle even after conservative treatment for 5 months. The pain developed after frequent ankle sprains and a splint was applied 1 year prior to her visit. For her medical history, she had received surgical treatment for neurofibromatosis 20 years ago. The pain intensity was 10/10 on the visual analogue scale (VAS), and the nature of the pain was squeezing, bursting and tingling. The patient showed allodynia and hyperalgesia in her left ankle and there was edema, skin color change and trophic change of the skin. We carried out various examinations including left ankle MRI, bone scan, autonomic nervous system function test, quantitative sensory test, electro-diagnostic study and diagnosed her as CRPS type 1.
After the CRPS diagnosis, we carried out various treatments including oral medications, lumbar sympathetic ganglion block, and common peroneal nerve block. After these treatments for 8 months, the edema and allodynia decreased, and the pain intensity was 4-6/10 on the VAS. Because there was some improvement in her symptoms, she did not come to our hospital and instead received treatment at an oriental herbal medicine clinic.
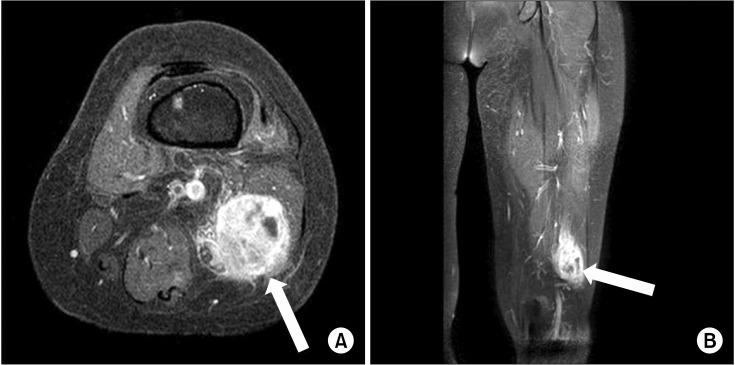
Six months later, she re-visited our pain clinic again because of aggravated pain in her left ankle. She complained of aggravated pain, numbness in her left ankle and difficulty in walking. Additionally, she stated there was a mass-like lesion in her left lower leg. Therefore, we conducted ultra-sonography, and there was a 2.7 × 2.7 × 4.3 cm mass with heterogeneous echogenicity at the posterior aspect of the left thigh (Fig. 1). Moreover, an electromyography and nerve conduction study (EMG-NCS) showed newly developed left sciatic neuropathy (more severely involved in the peroneal division than the tibial division), which had been normal 14 months prior to her visit. Left thigh MRI showed about a 3.0 × 3.1 × 5.2 cm sized mass along the common peroneal nerve in the left posterior thigh (Fig. 2), and US-guided biopsy diagnosed the mass as a malignant peripheral nerve sheath tumor. Positron emission tomography (PET) examination of the whole body was performed in order to rule out any possible metastasis and PET showed a lesion that was suggestive of lymph node metastasis in the thoracic region.

Therefore, she was transferred to an orthopedic and thoracic surgeon, and complete mass excision at the posterior thigh and mediastinal lymph node dissection were performed with video-assisted thoracic surgery. The durgical specimen containing a solid mass measuring 4.0 × 3.5 × 3.5 cm (Fig. 3) was proven to be MPNST. After the surgery, the numbness and pain decreased. And the patient received chemotherapy and radiation therapy.

Go to : 
DISCUSSION
MPNSTs are very rare sarcomas derived from the peripheral nerves or from the cells of the nerve sheath such as Schwann cells or perineural cells. It is called various names such as malignant schwannoma, neurogenic sarcoma, neurofibrosarcoma, and malignant neurilemmoma [5]. The age range for onset is from 20 to 50 years and MPNSTs rarely occurs in children. The incidence of MPNSTs is 0.001% in the general population and approximately 50% of MPNSTs occur in patients with NF 1 [1]. NF 1 is one of the cancer predisposing syndromes and MPNSTs associated with NF 1 generally have a poor prognosis. In our case, the patient had a past medical history of NF mass excision 20 years ago, which was related to the concurrent MPNST in the CRPS site. Moreover, MPNSTs are strongly related to a history of radiation exposure [6,7]. The most common anatomic sites for MPNSTs are the extremities, the trunk, and the head and neck, and they usually occur in the deep soft tissue, near a nerve trunk including the sciatic nerve, brachial plexus, and sacral plexus [1,3,4]. In our case, the MPNST occurred in the deep tissue of the left lower extremity and it was close to the common peroneal nerve. The major signs and symptoms of MPNSTs are palpable mass, pain, paresthesia, and hemiparesis, usually in a dermatomal pattern. If a patient does not have any specific signs and symptoms except for pain, an early diagnosis of MPNSTs is difficult. In our case, the patient suffered from complex regional pain syndrome type I in her left lower extremity for a long time. The mass was palpable in our case, but if the authors had not performed sonographic evaluation of her limb that was presenting pain, the diagnosis of MPNST would have been delayed. And if the diagnosis for MPNST was delayed, the tumor could have become bigger and the opportunity for total resection would have been lost.
Because a diagnosis of MPNST is very difficult, the taking of a patient's medical history and physical examination should be performed carefully. The medical history should include any neurogenic disorders or family history of neurogenic disorders and the physical examination should include any skin lesions and neurologic evaluations. CT and MRI may be useful in the diagnosis of MPNSTs. They show the extent of the lesion and distant metastasis if any and are therefore valuable in staging the lesion. Because hematogenous metastasis occurs most commonly in the lungs, high resolution chest CT should be performed on all patients with MPNSTs to evaluate for pulmonary metastasis [8]. Needle biopsy of the suspected cancerous mass is the most popular diagnostic study [9]. Electromyography may help in detecting localized tumors in the peripheral nerve [10].
Treatment of MPNSTs is similar to the treatment of other soft tissue sarcomas, and surgical resection is the first choice of treatment. The goal of surgical resection is complete resection of the tumor with histologically clear margins as the prognoses of patients with MPNSTs depend on a complete resection of the tumor [1]. Significant functional impairment may occur with a major nerve resection [1,9]. Although postoperative adjuvant therapy for MPNSTs is needed due to a higher local recurrence rate compared to other soft tissue sarcomas [11], the effect of the postoperative adjuvant therapy for MPNSTs is controversial. The effect of radiation therapy for MPNSTs has been proven by some studies [5,9,11]; however, other studies have failed to show a survival benefit of adjuvant radiation therapy [12]. Adjuvant chemotherapy in MPNSTs has been used in some cases [13]; however, the effect of chemotherapy is still controversial. In our case, MPNST was located in the deep tissue of the left lower extremity and the tumor was completely removed with preservation of the sciatic nerve. We are considering radiation therapy due to the risk of recurrence and pulmonary involvement.
The prognosis for patients with MPNSTs is poor due to local recurrence and metastasis. Local recurrence is more common than metastasis in patients with MPNSTs and the 5-year local recurrence rate has been reported as 27-42% in some studies [9,14-17]. The 5-year survival rate has been reported as 16-53% in several studies and histologically clear margin is the most important prognostic factor for the survival rate of the disease [1,5,10,11,14-17]. Therefore, complete surgical resection is most important in patients with MPNSTs. Other poor prognostic factors are tumors larger than 5 cm, a histological grade of 2 or 3, NF 1, and metastasis [18].
In conclusion, we think that if chronic pain does not respond well to proper treatment or symptoms suddenly change, clinicians must consider all the possibilities of musculoskeletal cancer or metastatic cancer in patients with CRPS.
Go to :
 XML Download
XML Download