

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
As a consequence of the rapidly aging population and the increasing prevalence of degenerative arthritis, there is a great demand on the drugs that manage inflammatory pain. Accordingly, selective inhibitors of cyclooxygenase (COX)-2 are one of the most widely used analgesics and its actions are well established to be mediated by the blockade of prostaglandin biosynthesis. However, several lines of evidence suggest that mechanisms of COX-2 inhibitor beyond the inhibition of COX and PG biosynthesis might also play an important role in their antinociception. Herrero and Headley. [1] reported that the opioid antagonist naloxone fully reversed or prevented the antinociception by flunixin, a non-steroidal anti-inflammatory drugs, in rats with carrageenan-induced inflammation of the hindpaw. Ibuprofen [2] and ketorolac [3] raised blood levels of endogenous opioids in human and rats, respectively. Pre-treatment with naltrexone diminished the analgesic effects of a COX-2 inhibitor, and its antinociception was abolished in rats made tolerant to the analgesic effects of morphine [4]. Taken together, these data indicate that there is a link between the opioid system and COX-2 inhibitor antinociception. However, the sites and mechanisms of any such connection are not yet clear.
The aim of this study was to clarify the role of opioid receptor subtypes on the effect of COX-2 inhibitor at the spinal level. Thus, µ, δ and κ opioid receptor antagonists were intrathecally administered to investigate the ability of opioid receptor subtype antagonists to reverse the antinociception induced by COX-2 inhibitor in the formalin test which shows an early phase of acute nociceptive response followed by a late phase response being related to more complex inflammatory reactions.
Go to : 
MATERIALS AND METHODS
All of the procedures were carried out with the approval of the Institutional Animal Care Committee, Research Institute of Medical Science. Male Sprague-Dawley rats weighing 250-300 g were used in these experiments. The rats were housed in a vivarium maintained at 20-23℃ with 12-h light/dark cycle and were given food and water ad libitum. A polyethylene tube (PE-10) was catheterized and inserted into the subarachnoid space in sevoflurane-anesthetized rats as described previously [5,6]. The rats were closely monitored and, if motor abnormalities appeared, they were euthanized through a volatile anesthetics overdose. Normal rats were kept in individual cages and a period of not less than 5 days was allowed for each rat to recover from intrathecal catheterization. Rats showing apparently normal behavior and weight gain were assigned to the experiment.
The following drugs were used in this study: DUP-697 (5-Bromo-2-(4-fluorophenyl)-3-[4-(methylsulfonyl)phenyl]-thiophene,), CTOP (d-Phe-Cys-Tyr-d-Trp-Orn-Yhr-NH2,), naltrindole (17-(cyclopropylmethyl)-6,7-dehydro-4,5α-epoxy-3,14-dihydroxy-6,7-2',3'-indolomorphian hydrochloride,) and GNTI (5'-guanidinyl-17-(cyclopropylmethyl)-6,7-dehydro-4,5α-epoxy-3,14-dihydroxy-6,7-2',3'-indolomorphian dihydrochloride, Tocris Cookson, Bristol, UK). Pharmacological characteristics of the above experimental drugs are presented in Table 1 [7-9]. All drugs were dissolved in dimethylsulfoxide (DMSO) and intrathecally administered using a hand-driven, gear-operated syringe in a volume of 10 µl solution followed by an additional 10 µl of saline to flush the catheter.
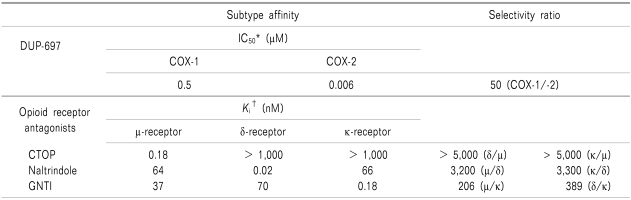
On experiment days, rats were placed in a restraining cylinder and held for 20 min for adaptation. To investigate the effect of COX-2 inhibitor in the formalin test, rats were treated with vehicle or DUP-697 (10, 30, 100, 300 µg), given 10 min before the formalin test. Doses of DUP-697 were determined by the maximum solubility and for approximately equal spacing on the log-scale. Rats were then pretreated with several opioid receptor antagonists in order to determine which subtypes of opioid receptor affected DUP-697 activity. These antagonists were administered intrathecally 10min before the delivery of intrathecal DUP-697 (300 µg). The formalin test was performed 10 min later. Three antagonists were selected on the basis of their selectivity on the receptor (Table 1) [7,9]. Doses of the opioid receptor antagonists were chosen based on previous experiment [10], in which the maximum dosage that did not affect the control formalin response or cause side effects such as motor impairment was determined. The opioid receptor antagonists used were as follows: µ opioid receptor antagonist, CTOP (15 µg); δ opioid receptor antagonist, naltrindole (10 µg); κ opioid receptor antagonist, GNTI (50 µg). Animals were tested only once. In total, 55 rats were tested in this study and the number of rats per group was 5-8.
For the formalin test, 50 µl of 5% formalin was injected subcutaneously into the plantar surface of the rat hindpaw. The number of flinches was counted for the 1-min periods at 1 and 5 min after the formalin injection, and every 5 min thereafter. Rats were observed for a total period of 60 min. Observed responses were divided into phase 1 (0-9 min) and phase 2 (10-60 min) of the formalin test. The researcher that tested the drugs was blind to the drug given to each animal. Data are expressed as means ± SEM. Time response data or dose-response data are shown either as the number of flinches or the percentage of control in two phases. Control study was done with DMSO, and the flinching number of the experimental group was converted to a percentage of control as follows:
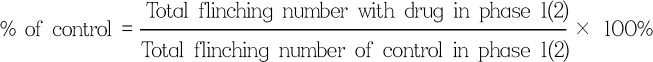
Dose-response data was analyzed using one-way analysis of variance (ANOVA) with Scheffe post hoc analysis. Comparison of antagonism for the effect of DUP-697 was analyzed by unpaired t-test. A P value < 0.05 was considered statistically significant.
Go to :
RESULTS
Subcutaneous injection of formalin into the paw evoked a biphasic pattern of flinching, with an early (phase 1) response lasting 5-10 min, and after a quiescent interval of 5-10 min, a subsequent late (phase 2) response up to 60 min. Fig. 1 shows the time course and dose-response data of intrathecal DUP-697, administered 10 min before formalin injection, for the formalin test. In the control group, total flinching number was (mean ± SEM) 28 ± 3 and 228 ± 15, during phase 1 and 2, respectively. Intrathecal DUP-697 reduced flinching response to 35-50% of the control group during phase 1 of the formalin test, but the extent of change was not statistically different over the range of administered dosage (Fig. 1B). During phase 2, DUP-697 suppressed the flinching response up to 48% of control in a dose-dependent manner (Fig. 1C).
 | Fig. 1Time course (A) and dose-response curves of intrathecal DUP-697 on flinching during phase 1 (B) and phase 2 (C) in the formalin test. DUP-697 was administered 10 min before the formalin injection. Data are presented as the number of flinches or the percentage of control. Each line represents means ± S.E.M. of 5-8 rats. Compared with control, *P < 0.05, †P < 0.005, ‡P < 0.001.
|
When CTOP was delivered intrathecally, 10 min before DUP-697 administration, total flinching number during phase 1 and 2 was 57% (P > 0.05) and 79% (P < 0.05) of the control value, respectively. Thus, pretreatment with µ opioid receptor antagonist CTOP reversed the antinociceptive effect of DUP-697 during phase 2, but not during phase 1, of the formalin test (Fig. 2). Total flinching number of the naltrindole-pretreated group during phase 1 and 2 was 73% and 74%, respectively (P < 0.05), and that of the GNTI-pretreated group was 69% and 76% of the control value, respectively (P < 0.05) (Fig. 2). Therefore, both δ and κ opioid receptor antagonists reversed the effects of DUP-697 in both phases.
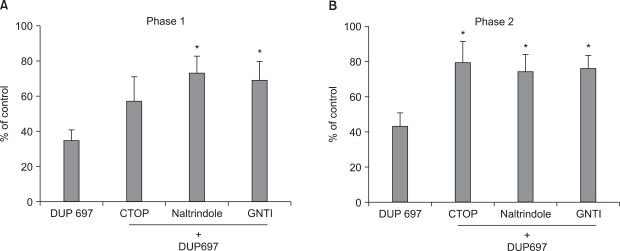 | Fig. 2The effects of intrathecal CTOP (15 µg), naltrindole (10 µg) and GNTI (50 µg) on the antinociception by intrathecal DUP-697 (300 µg) during phase 1 (A) and phase 2 (B) in the formalin test. CTOP, naltrindole and GNTI were administered 10min before the delivery of DUP-697, and then the formalin test was done 10 min later. Both of naltrindole and GNTI reversed the effect of DUP-697 during phase 1 and phase 2 in the formalin test. CTOP antagonized the antinociception of DUP-697 during phase 2, but not during phase 1. Data are presented as the percentage of control. Each bar represents means ± S.E.M. of 5-8 rats. Compared with DUP-697, *P < 0.05.
|
Go to :
DISCUSSION
It is generally thought that distinct mechanisms underlie the two phases of behavioral response in the formalin test. The phase 1 response is believed to represent a direct activation of sensory C fibers of primary afferent by formalin, thus phase 1 of the formalin test reflects acute pain. In contrast, the phase 2 response may result from the activation of wide dynamic range neurons with a continuously low level of activity in the primary afferent, thus representing a facilitated state [11].
In this study, intrathecal DUP-697 reduced the flinching response evoked by formalin injection during both phases. This finding suggests that this selective COX-2 inhibitor possesses a central mechanism of action, which is consistent with a previous report [12]. Moreover, pretreatment with intrathecal µ, δ and κ opioid receptor antagonists attenutated the effect of DUP-697, indicating that the endogenous opioid system mediate spinal antinociception of COX-2 inhibitor.
The involvement of the endogenous opioid system in the COX inhibitor analgesia has already been documented in other reports with various human and animal models. Troullos et al. [2] reported that ibuprofen enhances pituitary release of beta-endorphin by corticotroph cells in response to surgical stress in humans. In the mice model of nociception, intraperitoneal administration of naloxone significantly decreased the analgesic activity of ketorolac, suggesting that the opioid system might play a role in the COX inhibitor analgesia [13]. Recently, in a study by França et al. [4] selective inhibitors of COX-2 raised the nociceptive threshold above the normal non-inflamed level in a rat carrageenan model, and pre-treatment with naltrexone, an opioid receptor antagonist, abolished this effects. Moreover, in rats made tolerant to the anti-nociceptive effects of morphine, all antinociceptive effects of the COX-2 inhibitor were also abolished [4]. Taken together, these data indicate that there is a significant interaction between the opioid system and COX-2 inhibitor antinociception. However, until now, the roles of opioid receptor subtypes on the effect of COX-2 inhibitor at the spinal level were not determined.
In the current study, intrathecal CTOP, naltrindole, and GNTI attenuated the antinociceptive effect of intrathecal DUP-697 during both phases of the formalin test. However, the antinociception observed during phase 1 was antagonized by naltrindole and GNTI, but not CTOP. These observations suggest that δ and κ opioid receptors are involved in the activity of COX-2 inhibitor on the facilitated state as well as acute pain at the spinal level, whereas the µ opioid receptor is not related to the action of COX-2 inhibitors on acute pain.
The mechanism underlying opioid-mediated COX-2 inhibitor antinociception has not been clearly defined. Some COX inhibitors, such as paracetamol [14], have been reported to be able to bind to opioid receptors. However, it is unlikely that the COX-2 inhibitor used in this study, acted directly on the opioid receptor as an agonist because the nociceptive thresholds of the contralateral paw in inflamed rats were not affected, in contrast to the effects of the opioid receptor agonist, morphine [15]. In addition, the small effects the COX inhibitor had in rats with normal paws, were not reversed by a dose of naloxone high enough to block actions mediated at both the µ and κ opioid receptors [1,16]. A more likely explanation for the opioid-COX link would be the release of endogenous opioid peptides by the COX inhibitor, which is consistent with the increase of blood levels of endogenous opioids after COX inhibitor administration [2,3] and also compatible with the finding that prostaglandins can block endogenous opioid-mediated analgesia [17]. This possibility was further supported by the potentiation of celecoxib's effects by bestatin, a compound known to inhibit metabolism and consequent inactivation of endogenous opioid peptides [15]. On the other hand, the hyperalgesia, as a consequence of peripheral inflammation induced by a variety of agents, is associated with increased dynorphin expression [18-20], and opioid receptor antagonists reversed the decrease in dynorphin level induced by paracetamol [21]. Thus, some COX inhibitors may exert their antinociceptive effect also through the opioidergic system modulating dynorphin release in the central nervous system [21]. However, mechanisms of the unilateral analgesia, observed in the endogenous opioid-mediated COX inhibitor antinociception, remains to be further investigated, which may possibly be associated with inflammation-induced change in opioid receptor binding and G-protein coupling [22]. In addition, the differential role of the endogenous opioid system mediating COX inhibitor analgesia in the acute and facilitated states should be explored in future studies.
In conclusion, intrathecal administration of a COX-2 inhibitor decreased inflammatory pain, and its antinociceptive action was mediated by δ and κ opioid receptors in formalin-induced acute and facilitated pain. Additionally, the µ opioid receptor was involved in COX-2 inhibitor antinociception in the facilitated state.
Go to :
 XML Download
XML Download