

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The evidence that the development and persistence of neuropathic pain following a nerve injury is associated with inflammatory response has become clear [1,2]. Inflammatory mechanisms modulated by mediators facilitate not only primary afferent excitability but also synaptic transmission in the spinal cord. Such neuroinflammatory responses are more pronounced in traumatic neuropathy, such as peripheral nerve or spinal cord injury, than in other neurologic disease [2]. After peripheral nerve injury, the activation of microglia leads to rapid inflammation within the spinal cord, facilitating the development and maintenance of neuropathic pain [3]. Neuroinflammatory processes are regulated by various signaling pathways such as mitogen-activated protein kinase (MAPK), including p38 and c-Jun N-terminal kinase (JNK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), among others [4-7]. Signaling via the p38/JNK MAPK and NF-κB pathways is not only associated with the development of neuropathic pain but also the control of autophagy, which is essential for the balanced regulation of inflammation [6-8]. Autophagy is an ubiquitous cellular process required for homeostasis and survival, and initiated under stress conditions, such as malnutrition, organ damage, or inflammation [9]. Autophagy is also crucial for a balanced inflammatory response because of its regulatory role [9]. For spinal nerve ligation (SNL), it is known that the dysregulation of autophagy is related to the development of neuropathic pain [10]. A previous study showed that the modulation of MAPK/JNK-mediated autophagic processes decreases the release of the proinflammatory cytokines and effectively alleviates neuropathic pain after SNL [7].
Previously, the analgesic effects of Sec-O-glucosylhamaudol (SOG), isolated constituents from Saposhnikovia divaricata or Peucedanum japonicum, which is known as ‘Bangpung’ in Korea, were introduced [11-13]. The analgesic effect of SOG is thought to be related to an anti-inflammatory mechanism [11] and involve opioid receptors [12,13]. One recent study using RAW 264.7 cell lines, murine macrophage cells used for the study of inflammatory reactions, stimulated with lipopolysaccharide revealed that the anti-inflammatory effect of SOG is mediated by a reduction in proinflammatory cytokines through the inhibition of NF-κB activation and MAPK phosphorylation [14].
In the current study, we first evaluated the analgesic effect of SOG through behavioral tests and the involvement of the opioid receptor based on a naloxone challenge test. We hypothesized that SOG might have an analgesic effect in the rat SNL-mediated neuropathic pain model by decreasing the inflammatory response, concomitant with autophagy regulation, through the inhibition of p38/JNK MAPK or NF-κB pathways. The aim of this study was to investigate the effect intrathecal SOG on mechanical allodynia and the inflammatory process of neuropathic pain induced by SNL to confirm this hypothesis. Additionally, an in vitro study was conducted to examine the effect of SOG on the regulation of p38/JNK MAPK and NF-κB signaling pathways during the inflammatory process and autophagy induction.
Go to : 
MATERIALS AND METHODS
1. Animal preparation
The current study was approved by the Institutional Animal Care and Use Committee (CIACUC 2020-S0027) and followed the guidelines and ethical standards stipulated by the International Association for the Study of Pain [15]. The animal experiments were performed following the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines (https://www.nc3rs.org.uk/arrive-guidelines) for the investigation of experimental pain in animals. Specific pathogen-free male Sprague–Dawley rats (total n = 50; Damul Science, Daejeon, Korea) weighing 100-120 g were used for the experiments (Fig. 1). The rats were raised in cages located in a room with a constant temperature (20 to 23°C), with free access to food and water and a 12-hr light/dark cycle.
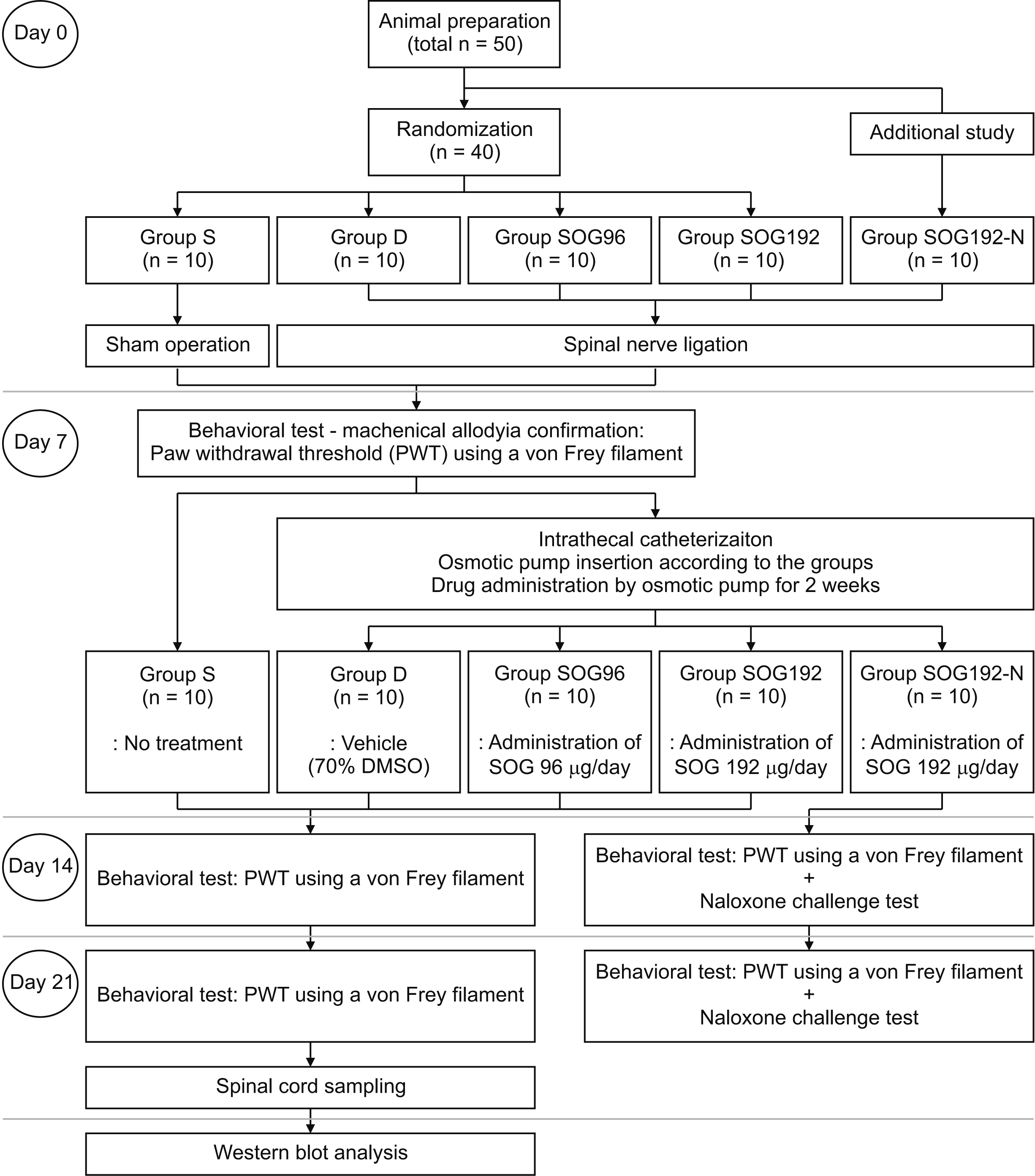 | Fig. 1Flow diagram illustrating the study protocol. Group S: sham-operated, Group D: control group treated with 70% dimethylsulfoxide (DMSO) after 7th day following spinal nerve ligation (SNL), Group SOG96: administered Sec-O-glucosylhamaudol (SOG) at 96 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL, Group SOG192: administered SOG at 192 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL, Group SOG192-N: administered SOG at 192 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL and subjected to the naloxone challenge test.
|
2. Randomization and grouping
Using computerized random numbers generation, rats were randomized into four groups (n = 10 in each group). Rats in group S were subjected to a sham operation. Meanwhile, other rats underwent SNL for the induction of neuropathic pain. The testing drugs were filled in the osmotic pump, according to group, and administered intrathecally for 2 weeks, continuously. Rats in group D were administered 70% dimethylsulfoxide (DMSO) only as a control. Those in group SOG96 and SOG192 were administered SOG at rate 96 and 192 μg/day, respectively, for 2 weeks. Those in group SOG192-N (n = 10) were administered SOG at 192 μg/day for 2 weeks but were also administered 2 mg/kg of intraperitoneal naloxone before assessment of the paw withdrawal threshold (PWT) for the naloxone challenge test.
3. Induction of neuropathic pain
Neuropathic pain was induced by segmental SNL according to the experimental model proposed by Chung et al. [16,17] for the experiment. After adaptation of naïve rats in the animal facility for 1 week, SNL was carried out in rats confirmed to have an absence of neurological abnormalities (n = 30). After the induction of general anesthesia with sevoflurane, incision of the skin on the midline of the L5-S2 spine was performed. The spine was exposed through dissection of the left paraspinal muscles, which were removed from the spinous processes. The left L5 and L6 spinal nerves were exposed by removing the transverse process of the L6 with a small rongeur. Then, each nerve was tightly ligated with a 6-0 silk at the distal site of the dorsal root ganglia. After suturing the incised wound, rats were observed in the cage until recovery from anesthesia to evaluate motor nerve damage. Only the rats without motor nerve damage were used for the study. Altogether, 10 rats received a sham operation without SNL. The PWT test using von Frey filaments was performed to confirm the development of neuropathic pain after 7 days. A flinching response of the hind paw during the application of a bending force of less than 4 g was set as the threshold for the development of neuropathic pain.
4. Intrathecal catheter insertion
After confirming the development of neuropathic pain, an intrathecal catheter was implanted under sevoflurane anesthesia in a stereotaxic apparatus. The skin of each rat was incised longitudinally on the level of the atlanto-occipital membrane to expose the cisterna magna. A polyurethane catheter (Rat Intrathecal Catheter®, NO 0007740; Alzet, Cupertino, CA) was inserted into the intrathecal space through the opening of the cisterna magna and was advanced caudally by 8.5 cm to place the proximal tip of the catheter on the level of the lumbar enlargement [18]. Intrathecal placement was confirmed by the leakage of cerebrospinal fluid through the distal tip of the catheter, which was anchored firmly by a suture. Then, the distal tip of the catheter was connected with the osmotic pump (ALZET micro-osmotic pump®, model ALZ-1002; pumping rate = 0.25 μL/h, fill volume = 0.5 mL, duration = 2 weeks; Alzet), which was filled with prepared drugs according to the groups. The osmotic pump was fixed in the subcutaneous tissue on the hind neck, and the skin was sutured with a 3-O silk. Rats were placed in individual cages for recovery from the anesthesia and their motor functions were observed. Only the rats without motor deficits were used in the study. The rats with motor deficits were euthanized immediately using an overdose of volatile anesthetics.
5. Drug and osmotic pump preparation
SOG (purity > 95%) was purchased from ChemFaces (Wuhan, Hubei, China). Diluted solutions of SOG were prepared at 16 and 32 μg/μL after dissolving in 70% DMSO. A concentration of DMSO greater than 70% was able to dissolve the SOG powder completely. The concentration of SOG was determined by a previous study, which showed an antinociceptive effect based on the formalin test [12]. In that study, the calculated 50% effective dose (ED50) was 48 µg, and it was decided to use twice the ED50 (96 µg) as a daily dose. Therefore, the dose was set as 96 µg/day for 2 weeks, initially, for the SOG96 group. The pumping rate of the osmotic pump was 0.25 μL/hr, and thus, the SOG was diluted to 16 μg/μL and filled the osmotic pump with 0.5 mL of diluted SOG for administration for 2 weeks. The osmotic pumps for group D were filled with 70% DMSO only as a control. Those for group SOG96 were filled with SOG at a concentration of 16 μg/μL, whereas those in group SOG192 and SOG192-N were filled with SOG at a concentration of 32 μg/μL.
6. Assessment of mechanical allodynia
To assess the development of neuropathic pain and mechanical allodynia, PWT using von Frey filaments (Stoelting, Wood Dale, IL) was assessed with a 7-day interval until 3 weeks after SNL. The PWT was calculated by the up and down method [19]. The rats were placed in the cage with a mesh floor and adapted to the laboratory environment for 30 minutes. Then, the mechanical stimulation was applied with a series of eight von Frey filaments (0.4, 0.7, 1.2, 2.0, 3.6, 5.5, 8.5, and 15 g) according to the up and down method. A vertical mechanical stimulation for 5 seconds was applied to the plantar surface of the left hind paw, exposed via apertures in the mesh floor. Responses such as characteristic flinching of the hind paw or abrupt withdrawal during the stimulation with a filament were regarded as a positive response. The cut-off value was a negative response to 15 g. Each filament was applied twice with a 3 minutes resting period for response measurements.
7. Naloxone challenge test
To determine the association between SOGs and opioid receptors, a further experiment using the naloxone challenge test was conducted based on the SOG192-N group [20]. The rats from group SOG192-N were treated and administered SOG in the same way as for group SOG192. After the basal assessment of PWT on the 14th and 21st day, a 2 mg/kg dose of naloxone (Tocris Cookson, Avonmouth, UK) was administered intraperitoneally. Then, the rats were placed in the cage and the PWT was measured after 30 and 60 minutes. Further, the signs of opioid withdrawal such as spontaneous vocalization, head shakes, or escape attempts were observed for 1 hour [20].
8. Spinal cord sampling
On the 21st day, rats were anesthetized with a sevoflurane overdose and euthanized by decapitation. Then, ice-cold phosphate-buffered saline was flushed from the caudal end of the vertebral column for the isolation of the spinal cord. The spinal cord was cut at the L4-L6 level and the ipsilateral dorsal spinal cord was obtained. Immediately, the obtained tissue sample was stored at −70°C with liquid nitrogen.
9. Western blotting
The tissue samples from the frozen specimens were dissected and homogenized in lysis buffer with a Dounce homogenizer. Samples were lysed using radioimmunoprecipitation assay buffer with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO). The lysed tissues were centrifuged at 10,000 × g for 20 minutes for clarification. After quantification of the protein content, the lysates were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis in 12%-15% acrylamide gels. Immunoblotting was conducted after transferring the lysate to polyvinylidene difluoride membranes (Millipore, Billerica, MA). The corresponding antibodies were as follows: anti-rabbit polyclonal atg8/LC3 antibody, SQSTM1/p62 antibody, and Beclin 1 antibody, obtained from Cell Signaling (Irvine, CA); phospho-JNK, p38, and p65, obtained from Cell Signaling (Danvers, MA); interleukin-1 (IL-1), interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α), and β-actin (sc-70,319), purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Chemiluminescence Western Blotting Detection Reagents (Millipore) were used for the detection of bands, and the quantification of blots was conducted with ImageJ densitometry software (National Institutes of Health, Bethesda, MD) for densitometric analyses.
10. Cell culture for in vitro experiments
To evaluate the effect of SOG on the p38/JNK MAPK pathway and autophagy induction in capsaicin-exposed cells, additional experiments were conducted. The rat neonatal dorsal root ganglion (DRG) neurons (Cat. #R-DRG-505; Lonza Inc., Houston, TX) were maintained in PNGMTM primary neuron growth medium (BulletKitTM, CC-4461; Lonza Inc.) in a 95% air/5% CO2 humidified incubator (at 37°C), with the addition of heat-inactivated 10% fetal bovine serum, 50 μg/mL penicillin, and 50 μg/mL streptomycin. The cells were treated with capsaicin (Sigma-Aldrich) at different concentrations (0, 50, 100, 200, and 400 μM), and SOG (30 μg) was added to the cells treated with capsaicin at 400 μM for to evaluate its effects. Next, to determine whether the effect of SOG on cytokine and autophagy was associated with the p38/JNK MAPK pathway, pharmacological inhibitors (SB203580, p38 inhibitor, 20 μM; SP600125: JNK inhibitor, 20 µM; Sigma-Aldrich) were used, and the results were analyzed by western blot analysis using appropriate antibodies.
11. Statistical analysis
The sample size was calculated based on the general considerations for sample size estimation in an animal study using “G*Power 3.1” software [21]. According to the results of a previous study, which showed the significant antinociceptive effect as a 70.6% reduction in the flinching response after the administration of SOG at 30 μg intrathecally [12], the effect size was calculated as 0.519. With α = 0.05, 85% power, and a correlation of 0.5 for three consecutive PWT tests based on repeated measures analysis of variance, the total sample size was calculated as 36 in four groups (except for the naloxone challenge test). It was decided to make the sample size for each group of 10 given a drop-out rate of 10%. For additional evaluation based on the naloxone challenge test, the total sample size was decided to be 50. Data are expressed as the mean ± standard error of the mean. The results of mechanical allodynia using the PWT were analyzed by a repeated measures two-way analysis of variance or Kruskal–Wallis test followed by Scheffe’s post-hoc test. According to Mauchly’s test of sphericity, data did not satisfy the spherical assumption (P < 0.001), and thus, the corrected values, using Greenhous–Geisser, were used for the statistical analysis. Then, the differences between groups were analyzed by a Mann–Whitney U-test for multiple comparisons. A t-test was performed for comparisons of densitometry between the control and experimental groups. A P value of less than 0.05 was considered statistically significant.
Go to :
RESULTS
1. Continuous administration of intrathecal SOG increases the PWT
Mechanical allodynia was confirmed based on a PWT of less than 1 g in all rats except for those in group S (sham-operated) 7 days after the SNL procedure. The PWT was significantly increased in rats from groups SOG96 and SOG192 compared with those in Group D (P < 0.001) after administration of testing drugs during the observational periods. There were no significant differences in the PWT among the groups on the 7th day after SNL (Fig. 2). The rats from groups SOG96 and SOG192 showed a significantly higher PWT than those from group D on the 14th and 21st day after SNL (P < 0.001). However, there were no significant differences in the PWT between group SOG96 and SOG 192 on the 7th and 21st day after SNL (P = 0.728 on the 7th day; P = 0.089 on the 21st day). These results indicate that the continuous administration of intrathecal SOG decreased mechanical allodynia induced by SNL, but had a ceiling effect.
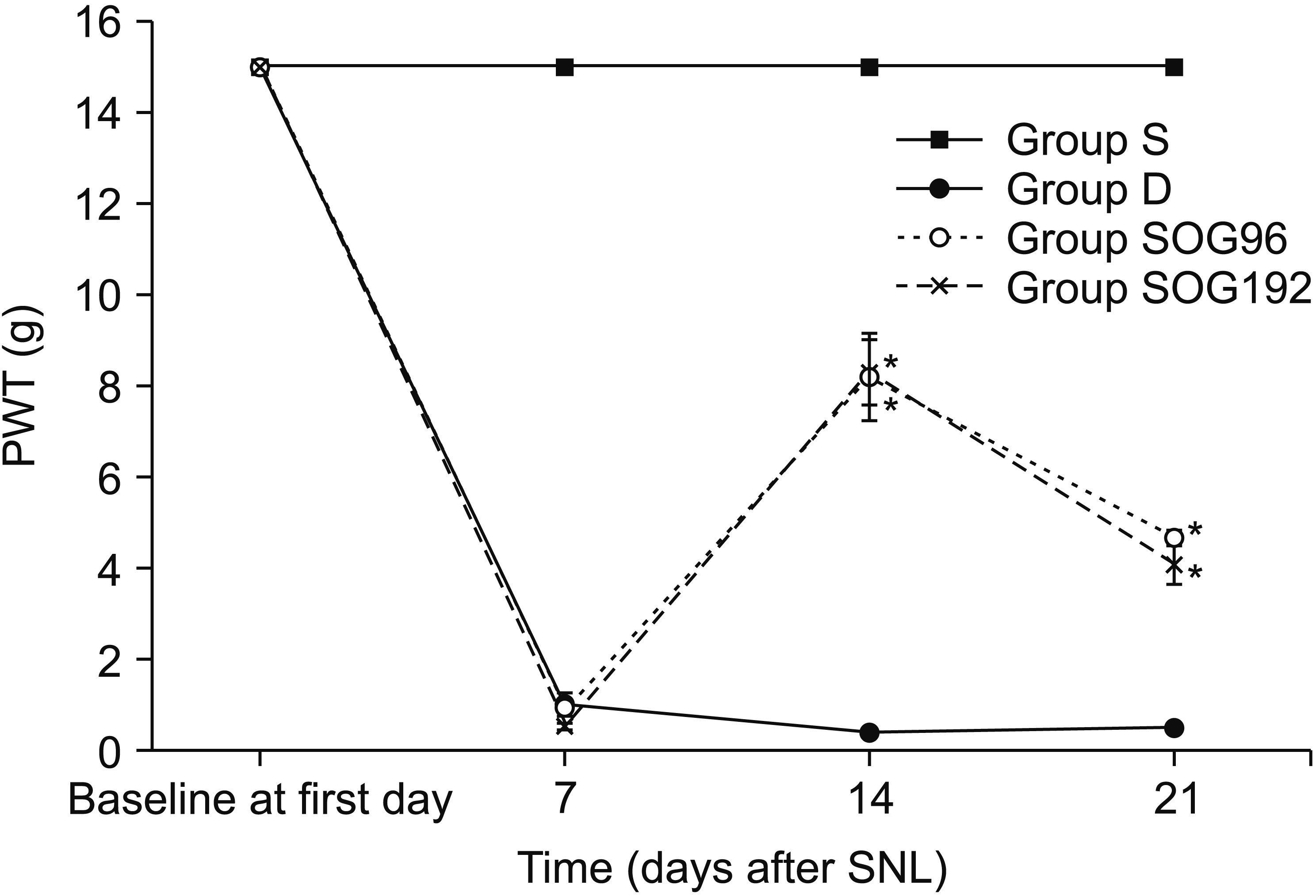 | Fig. 2Paw withdrawal threshold (PWT) during observational periods. The PWT was observed on the 7th, 14th, and 21st day after spinal nerve ligation (SNL). On the 7th day after SNL, mechanical allodynia was confirmed in all rats based on a significant decrease in PWT compared to that in group S (P < 0.001). There were no significant differences among experimental groups including groups D, SOG96, and SOG192. The rats treated with continuous administration of Sec-O-glucosylhamaudol (SOG) intrathecally (group SOG96 and group SOG192) showed a higher PWT than those in the control group (group D) on the 14th and 21st day after SNL (P < 0.001). There were no significant differences in the PWT between groups SOG96 and SOG192 during the observational periods (P = 0.728 on 7th day; P = 0.089 on 21st day). Group S: sham-operated, Group D: control group treated with 70% dimethylsulfoxide after 7th day following SNL, Group SOG96: administered SOG at 96 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL, Group SOG192: administered SOG at 192 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL. *P < 0.05 compared with group D.
|
2. Naloxone challenge test
An additional experiment using the naloxone challenge test was conducted to determine the association between SOG and opioid receptors. Intraperitoneal administration of naloxone did not alter the PWT at either 30 or 60 minutes on either the 14th or 21st day after SNL (Fig. 3). Moreover, no signs of opioid withdrawal were observed until 60 minutes. According to these results, the analgesic effect of intrathecal SOG on the mechanical allodynia induced by SNL would not be related to opioid receptors.
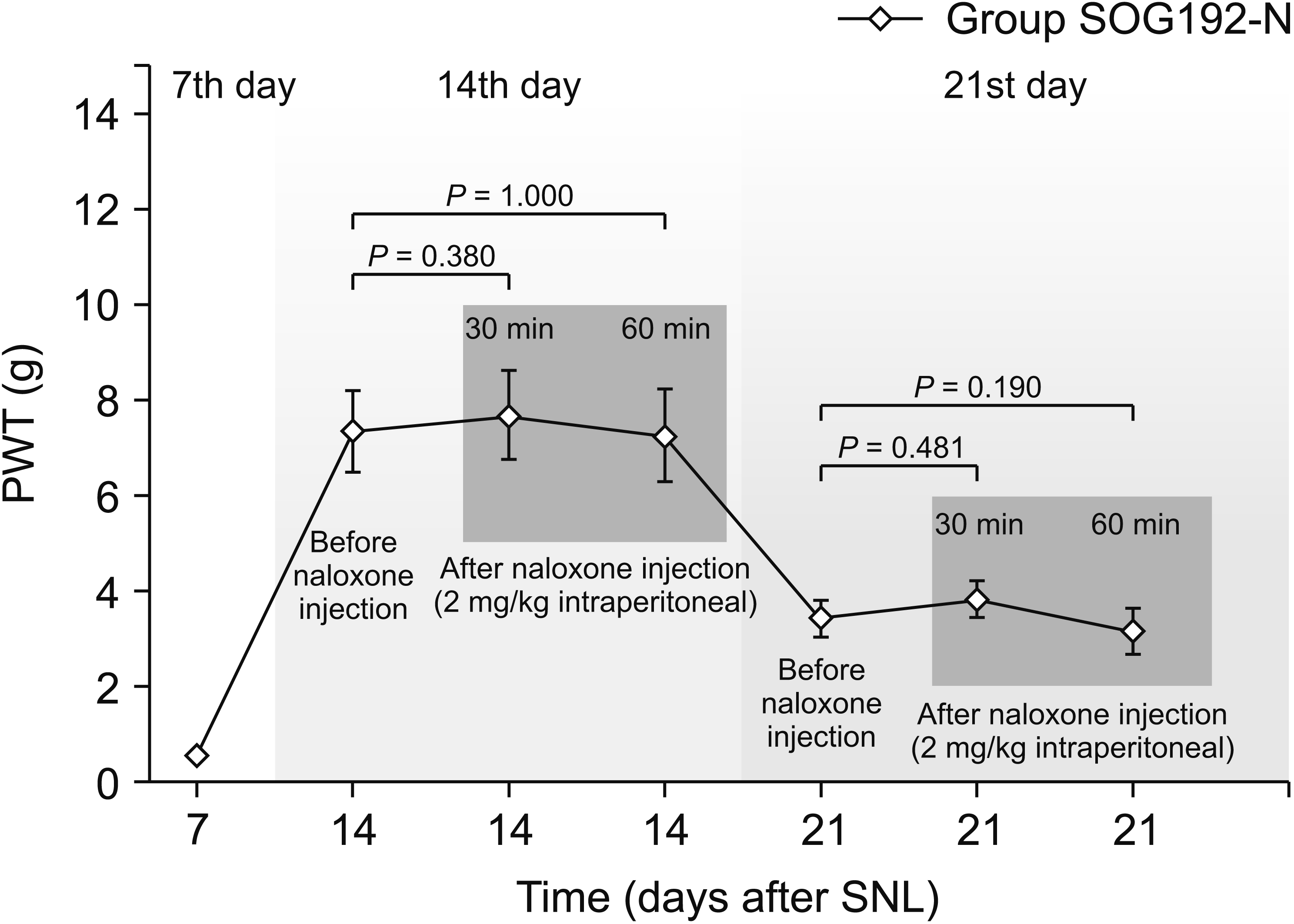 | Fig. 3Naloxone challenge test on the 14th and 21st day. The paw withdrawal threshold (PWT) after spinal nerve ligation (SNL) did not change after the intraperitoneal administration of naloxone (2 mg/kg) either at 30 or 60 minutes. No signs of opioid withdrawal were observed until 60 minutes. Group SOG192-N: administered Sec-O-glucosylhamaudol at 192 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL and subjected to the naloxone challenge test.
|
3. p38/JNK MAPK, NF-κB, proinflammatory cytokines, and autophagic activation in SOG-mediated pain
We next investigated the effect of SOG on the p38/JNK MAPK and NF-κB pathways, proinflammatory cytokines, and autophagy activation on the 21st day after SNL, with an in vivo experiment, via western blotting and densitometry (Fig. 4). The p38/JNK MAPK pathway, which is related to the development and maintenance of neuropathic pain [22], was examined. SNL in the experimental groups resulted in an increase in the activation of p38 MAPK and phosphorylated JNK-I/II compared to the levels in group S (p38 MAPK: P = 0.004, phosphorylated JNK-I: P < 0.001, phosphorylated JNK-II: P = 0.001, Fig. 4A). The expression levels of p38 and JNK were markedly decreased upon SOG treatment in group SOG96 and SOG192 compared with those in group D (both P < 0.001). The expression levels of p65 NF-κB, implicated in the production of IL-1β and TNF-α in the development of neuropathic pain [6], were also increased after SNL in the experimental groups (p65 NF-κB: P < 0.001, Fig. 4A). However, the expression level of NF-κB was significantly decreased by SOG treatment in groups SOG96 and SOG192, as compared to that in group D (P = 0.015 and P < 0.001, respectively). The production of proinflammatory cytokines such as IL-1, IL-6, and TNF-α was increased after SNL (IL-1: P = 0.001, IL-6: P = 0.001, TNF-α: P = 0.01, Fig. 4A). However, the expression level of IL-1 was not increased in group SOG192 compared to that in group S (P = 0.571). Especially, the expression levels of IL-1 and TNF-α, which play a pivotal role in neuropathic pain [23,24], were also markedly decreased by SOG treatment in groups SOG96 and SOG192 compared to those in group D (both P < 0.001), but there was no significant decrease in the levels of IL-6. These results suggest that SNL-mediated activation of the p38/JNK MAPK and NF-κB signaling pathways was inhibited by SOG.
 | Fig. 4Expression of mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinase (JNK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and proinflammatory cytokines in the ipsilateral dorsal horn of the L5 spinal cord after spinal nerve ligation (SNL) on the 21st day (A). The expression levels of p38 MAPK, phosphorylated JNK-I/II, and p65 NF-κB were increased after SNL and markedly decreased by treatment with Sec-O-glucosylhamaudol (SOG). The production of proinflammatory cytokines was increased after SNL. SOG markedly decreased the expression levels of IL-1 and TNF-α, but there was no significant decrease in the levels of IL-6. Autophagic activation in the ipsilateral dorsal horn of the L5 spinal cord on the 21st day after SNL is shown (B). Upregulation of autophagy by SNL was confirmed by the increased expression levels of microtubule-associated protein 1 light chain 3 (LC3)-I/II and Beclin 1 with a concomitant decrease in p62, and autophagy processes were downregulated after SOG treatment. Values were normalized to β-actin levels. The error bars indicate mean ± standard deviation. IL-1: interleukin-1, IL-6: interleukin-6, TNF-α: tumor necrosis factor-alpha, Group S: sham-operated, Group D: control group treated with 70% dimethylsulfoxide after the 7th day following SNL, Group SOG96: administered SOG at 96 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL, Group SOG192: administered SOG at 192 μg/day for 2 weeks using an osmotic pump after the 7th day following SNL. aP < 0.05 vs. Group S. bP < 0.01 vs. Group S. cP < 0.05 vs. Group D. dP < 0.01 vs. Group D.
|
Autophagic activation by SNL was evaluated by densitometric analysis of western blot results (Fig. 4B). The increased autophagic activation mediated by SNL was confirmed by the increased expression levels of LC3-I/II and Beclin 1 with a concomitant decrease in p62 (SQTSMI/sequestosome 1, an autophagy adaptor molecule) (LC3-I: P = 0.002, LC3-II: P < 0.001, Beclin 1: P = 0.002, p62: P < 0.001). After SOG treatment, the SNL-mediated increase in the expression levels of LC3-I/II and Beclin 1 decreased (both P < 0.001) and p62 levels increased (both P < 0.001). These results indicate that SNL-mediated autophagy was inhibited by SOG.
4. SOG suppresses capsaicin-induced autophagy by decreasing p38/JNK MAPK and NF-κB in vitro
To evaluate the cellular nociceptive mechanism, capsaicin, which is involved in pain sensation, was used [25]. Capsaicin treatment induced an increase in phosphor-JNK and phosphor-p38 in R-DRG-505 cells (Fig. 5A). The expression levels of p38/JNK MAPK and NF-κB were increased in a dose-dependent manner after capsaicin treatment, and maximal effects were shown with 400 μM (data are not shown). Furthermore, capsaicin induced an increase in proinflammatory cytokines, including IL-1β, IL-6, and TNF-α. Consequently, the upregulation of autophagy induced by capsaicin was confirmed based on the changes in LC3-I/II, p62 protein, and Beclin 1 (Fig. 5B). Specifically, the expression levels of LC3-I/II and Beclin 1 were increased, whereas the expression of p62 was decreased. These effects were mitigated by SOG treatment. The expression levels of phospho-p38 and phosphor-JNK MAPK, as well as NF-κB, were reduced with a decrease in proinflammatory cytokines, including IL-1β and TNF-α, but not IL-6. Especially, the levels of p38 MAPK were decreased markedly after SOG treatment. Moreover, the downregulation of autophagic processes was confirmed by the decrease in LC3-I/II and Beclin 1 and increase in p62 protein.
 | Fig. 5Expression of mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinase (JNK), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and proinflammatory cytokines in capsaicin-exposed R-DRG-505 cells. Capsaicin treatment induced an increase in the expression levels of p38/JNK MAPK, p65 NF-κB, IL-1β, IL-6, and TNF-α (A) with the concomitant upregulation of autophagy (B), which were reversed by Sec-O-glucosylhamaudol (SOG) treatment. Pharmacological p38/JNK MAPK inhibitors resulted in the consequent inhibition of p65 with decreased expression of proinflammatory cytokines and the downregulation of autophagy. Values were normalized to β-actin levels. The error bars indicate mean ± standard deviation. SB203580: p38 inhibitor, SP600125: JNK inhibitor, IL-1: interleukin-1, IL-6: interleukin-6, TNF-α: tumor necrosis factor-alpha, LC3: microtubule-associated protein 1 light chain 3. aP < 0.05 vs. Capsaicin 0 μM. bP < 0.01 vs. Capsaicin 0 μM. cP < 0.05 vs. Capsaicin 400 μM. dP < 0.01 vs. Capsaicin 400 μM.
|
Additionally, the authors examined whether activation of the p38/JNK MAPK signaling pathway was involved in autophagy induction in capsaicin-exposed cells. For this purpose, a pharmacological p38 inhibitor (SB203580) and JNK inhibitor (SP600125) were used. The inhibition of p38 MAPK resulted in p65 inhibition and a reduction in the expression levels of IL-1β and TNF-α, but this had no effect on IL-6. The p38 inhibitor also resulted in the downregulation of autophagy processes with a decreased LC3-I level and increased p62 level. Meanwhile, JNK inhibition mainly reduced the expression levels of IL-6 and markedly suppressed the capsaicin-induced autophagy levels.
Go to :
DISCUSSION
In the current study, the development of neuropathic pain after SNL in rats was confirmed by a decrease of PWT. With the increase in the PWT, the expression of the p38/JNK MAPK pathway and NF-κB increased, leading to an increase in the expression of proinflammatory cytokines with autophagy activation. The continuous administration of intrathecal SOG via an osmotic pump decreased mechanical allodynia induced by SNL. The activation of signaling pathways was inhibited, which led to a decrease in the expression of proinflammatory cytokines with the concomitant downregulation of autophagy. Those results indicate that SOG ameliorates SNL-induced neuropathic pain by regulating proinflammatory cytokines and downregulating of autophagy through the inhibition of the p38/JNK MAPK and NF-κB signaling pathways.
Previous studies have shown the analgesic effect of SOG on various pain models, which was determined to be associated with an anti-inflammatory effect, COX inhibition, or opioid receptor interactions [11-13,26]. Moreover, as mentioned, a recent study demonstrated the anti-inflammatory effects of SOG on inhibiting the transcription of proinflammatory cytokines via the suppression of NF-κB activation and MAPK phosphorylation [14]. Those studies suggested the therapeutic potential of SOG in connection with inflammatory regulation.
In terms of neuroinflammatory mechanisms, the development of neuropathic pain after peripheral nerve injury is associated with immense inflammatory cascades via the activation of glial cells and astrocytes in the DRG, which leads to the release of proinflammatory cytokines [1,27-29]. Such neuroinflammatory processes, after nerve injury, are regulated by various signaling pathways, such as p38 MAPK, JNK, and NF-κB, among others [4-7]. In stressful conditions such as spinal cord injury, the stress-activated protein kinase group of MAPKs, including JNK and p38 MAPK, which play an important role in the development of neuropathic pain via the production of IL-1β and TNF-α, are activated [30]. p38 MAPK is implicated in the inflammatory responses with microglial activation and the production of inflammatory cytokines, whereas JNK mediates apoptosis of oligodendrocytes, which leads to neuronal degeneration or dysfunction. NF-κB is the primary regulator of inflammatory responses, such as the upregulation of proinflammatory cytokines like IL-1β, IL-6, and TNF-α [31]. NF-κB is associated with chronic inflammation during the development of neuropathic pain [5]. Therefore, the inhibition of NF-κB could be a potential strategy for functional recovery following spinal cord injury [32]. Previous studies confirmed the increase in the expression of p38/JNK MAPK and NF-κB signaling pathway components after SNL [6,7]. In the current study, an increase in the expression of p38 MAPK, JNK, and NF-κB after SNL were identified, and the increase in proinflammatory cytokines through the activation of signaling pathways was confirmed.
Autophagy is a ubiquitous cellular process required for homeostasis and survival that functions through a lysosomal degradation pathway [33]. Autophagy is activated during stressful situations such as starvation, hypoxia, organ damage, or inflammation and is necessary for the elimination and recycling of harmful components in damaged cells [3,9,33]. Although the mechanism of autophagy is very complicated and varies according to the organs, injury models, contexts, and times, it primarily plays a protective role in the development of neuropathic pain [3]. However, the excessive induction of autophagy might lead to type II programmed cell death. After SNL, increased activation of autophagy contributes to excessive inflammatory processes in the spinal cord via the activation of microglial cells and the aggravation of neuropathic pain [3,7,34].
The regulatory pathways of autophagy and proinflammatory cytokines interact with each other [33]. As the cytokines, induced by signaling pathways and autophagy, regulate each other, the activation and regulation of autophagy are closely associated with MAPK and NF-κB [9,33]. After peripheral nerve injury, intracellular molecular signaling pathways, such as MAPK and NF-κB, are activated and the production of IL-1β and TNF-α is increased. Autophagy is also activated by IL-1β and TNF-α through modulation by JNK and p38 MAPK-NF-κB [9]. In particular, TNF-α, which is involved in the pro-inflammatory processes related to the development of neuropathic pain, positively regulates autophagy and increases the expression of LC3 and Beclin 1 through activation of the p38/JNK MAPK signaling pathway. Meanwhile, autophagy modulates the induction of TNF-α in a context-dependent manner [7,9].
Autophagic activity can be confirmed by changes in marker proteins [7,35]. LC-3 is a reliable marker protein for autophagic activity, and Beclin 1 is a key protein in the induction of autophagy. SQSTM1/p62 is an autophagy adaptor molecule that binds to LC3 and is degraded in autolysosomes. According to the results of the current study, increased activity of autophagy after SNL was confirmed by increased expression levels of LC3-I/II and Beclin 1 proteins and a decrease in the expression of SQSTM1/p62, which was reversed by treatment with continuous intrathecal SOG administration. These results indicate that the inhibition of autophagy induction can be mediated by SOG.
To confirm the mechanism through which SOG regulates proinflammatory cytokines and autophagy, mediated by intracellular signaling pathways, an additional in vitro study was conducted using R-DRG-505 cells. The authors focused on p38/JNK MAPK pathway activation with respect to its involvement in autophagy, and capsaicin was used to activate these signaling pathways and autophagy as a pain sensation-inducer [7]. Similar to the results shown after SNL in vivo, capsaicin increased the expression of p38, JNK, NF-κB, and proinflammatory cytokines with the upregulation of autophagy in DRG cells in vitro. Such increases were reversed by treatment with SOG. Additional experiments with a p38 inhibitor and JNK inhibitors showed the decreased expression levels of proinflammatory cytokines in capsaicin-exposed cells. JNK inhibition mainly reduced the expression levels of IL-6 and downregulated LC3-II. Furthermore, p38 inhibition resulted in reduced expression of IL-1β and TNF-α, but not IL-6, which led to the downregulation of autophagy with a decrease in LC3-I and increase in p62. These results indicated that the p38/JNK MAPK pathway might act on inflammatory responses via different pathways. Given that the expression patterns of the signaling pathways and cytokines with SOG were similar to those with the p38 inhibitor, the mode of action of SOG is thought to be primarily related to p38 MAPK with the support of JNK. Moreover, the inhibition of p65 NF-κB, which is involved in the production of IL-1β and TNF-α in the development of neuropathic pain, was regulated by the modulation of p38 [6]. Collectively, the effect of SOG on the suppression of inflammatory responses might occur via inhibition of the p38/JNK MAPK pathway, and SOG could have a stronger effect on p38 MAPK. Furthermore, these results suggested that autophagy might be associated with the potential effects of SOG on the suppression of inflammatory responses.
There are several limitations to this study. First, the role of autophagy in the development of neuropathic pain after SNL and the therapeutic target in the regulation of autophagy is unclear because of its complex mechanism. In the current study, the upregulation of autophagy was confirmed by the increase in LC3 with a concomitant decrease in p62, which was thought to underlie the development of neuropathic pain, and autophagy suppression was a part of the analgesic mechanism of SOG. However, a previous study revealed the dysregulation of autophagy with an increase in both LC3 and p62 (rather than upregulation), which was the major findings associated with autophagy-induced neuropathic pain [10,35]. The main reason for this difference was thought to be the time from SNL (21 days vs. 7-14 days) and species differences (Sprague–Dawley rat vs. mouse). However, it is clear that autophagy activity increased after SNL compared to basal activity, given the increased expression levels of LC3, which represents the upregulation of autophagy as a response to stress. To overcome this limitation, further research at different times with morphologic evaluations is required. Second, additional research using different doses of SOG or infusion rates is needed. Interestingly, a previous study showed the opioid receptor-associated analgesic effect of SOG [13], but the effect of SOG was not related to opioid receptors according to the naloxone challenge test in this study. According to the result of the current study, intraperitoneal naloxone did not reverse the antiallodynic effect of SOG. This discrepancy might have been caused by a difference in receptor occupancy according to the dosage. Although we determined the concentration of SOG according to the ED50 for the analgesia based on a previous study [12], the dosage used in previous studies was for a single bolus administration (maximal 100 µg, once), whereas continuous administration with an osmotic pump (dosages of 96 µg and 192 µg during 24 hours) was used in this study. Therefore, the intrathecal concentration of SOG would be lower in the current study than the previous study. These differences in dosage and administration methods could cause differences in the intrathecal concentration and effectiveness of SOG.
In conclusion, this study demonstrated that p38/JNK MAPK and NF-κB signaling pathways induce an increase in the expression of proinflammatory cytokines with the concomitant upregulation of autophagy with SNL-induced neuropathic pain. The continuous administration of intrathecal SOG inhibited the induction of signaling pathways with a reduction in proinflammatory cytokines and the downregulation of autophagy. Consequently, SOG alleviated mechanical allodynia, and its mechanism is thought to be related to the regulation of the p38/JNK MAPK and NF-κB signaling pathways associated with autophagy during the neuroinflammatory processes following SNL.
Go to :
 XML Download
XML Download